 By Ivana Melo Pereira
By Ivana Melo Pereirasexta-feira, 21 de novembro de 2008
Pelagra e pele
Pequeno trecho do artigo Pellagra and skin, o objetivo é enfatizar os sintomas da pelagra, principalmente o "colar de casal".



Fonte:
Review Pellagra and skin
Kaliaperumal Karthikeyan, MD, and Devinder Mohan Thappa, MD, DHA, MNAMS
From the Department of Dermatology and STD, Jawaharlal Institute of
Postgraduate Medical Education and Research, Pondicherry – 605 006, India


Epidemiology of pellagra
Pellagra has occurred in many geographic areas as one of the commonest world-wide afflictions.
Once a formidable and widespread deficiency disease among a malnourished population subsisting mainly on maize diets, pellagra has declined in all parts of the world. Although today the condition is rather uncommon, occasional cases are found. It is still endemic in remote areas of the world where green vegetables, fruit, and animal proteins are unobtainable. Pellagra was endemic in Egypt until the gradual replacement of maize with wheat brought about improvements. In areas of Africa, south of the Sahara, namely in Nyasaland (Malawi) and Basutoland (Lesotho), as well as in areas of South Africa where maize is still a basic food, pellagra continues to be a problem.
The disease is particularly common in areas populated by the Bantu tribe in South Africa and is
endemic in a few areas of Western Africa, notably Angola. Besides Africa, a significant number of cases have been reported in Asia.
The disease is endemic in the deltas of some rivers in China. However, most reports related to pellagra in Asia have come from India. While pellagra is historically a disease of a maize-eating population, it has been reported in the Telangana area of Andhra Pradesh in India and in some
segments of the population who eat a cereal known as jowar (Sorghum vulgare); these people consume very little milk or other foods of animal origin.
Amino-acid imbalance caused by an excess of leucine is the cause of pellagra in both jowar and maize eaters. Excess of leucine appears to interfere in the conversion of tryptophan to niacin. Pellagra is an endemic disease among the population of India whose basic diet is maize or jowar (Sorghum vulgare).
A striking fall in the incidence of pellagra occurred in the USA following the discovery of nicotinic acid’s role; nowadays it is rarely encountered. In the European countries it appears nowadays sporadically.
Pellagra has occurred in many geographic areas as one of the commonest world-wide afflictions.
Once a formidable and widespread deficiency disease among a malnourished population subsisting mainly on maize diets, pellagra has declined in all parts of the world. Although today the condition is rather uncommon, occasional cases are found. It is still endemic in remote areas of the world where green vegetables, fruit, and animal proteins are unobtainable. Pellagra was endemic in Egypt until the gradual replacement of maize with wheat brought about improvements. In areas of Africa, south of the Sahara, namely in Nyasaland (Malawi) and Basutoland (Lesotho), as well as in areas of South Africa where maize is still a basic food, pellagra continues to be a problem.
The disease is particularly common in areas populated by the Bantu tribe in South Africa and is
endemic in a few areas of Western Africa, notably Angola. Besides Africa, a significant number of cases have been reported in Asia.
The disease is endemic in the deltas of some rivers in China. However, most reports related to pellagra in Asia have come from India. While pellagra is historically a disease of a maize-eating population, it has been reported in the Telangana area of Andhra Pradesh in India and in some
segments of the population who eat a cereal known as jowar (Sorghum vulgare); these people consume very little milk or other foods of animal origin.
Amino-acid imbalance caused by an excess of leucine is the cause of pellagra in both jowar and maize eaters. Excess of leucine appears to interfere in the conversion of tryptophan to niacin. Pellagra is an endemic disease among the population of India whose basic diet is maize or jowar (Sorghum vulgare).
A striking fall in the incidence of pellagra occurred in the USA following the discovery of nicotinic acid’s role; nowadays it is rarely encountered. In the European countries it appears nowadays sporadically.

Fonte:
Review Pellagra and skin
Kaliaperumal Karthikeyan, MD, and Devinder Mohan Thappa, MD, DHA, MNAMS
From the Department of Dermatology and STD, Jawaharlal Institute of
Postgraduate Medical Education and Research, Pondicherry – 605 006, India
Por: Pollana Roberta Alves Campos
RESUMÃO
" Do italiano pelle agra, pele áspera, veio o termo pelagra para designar uma patologia caracterizada por dermatose que acometia povos assumidamente grandes consumidores de milho, como os italianos e os norte-americanos. Mais uma vez foi Funk que ligou o problema a uma deficiência alimentar que poderia ser evitado com o consumo de leite, ovos e carne.
Goldberger, outro grande estudioso das vitaminas conseguiu, usando dietas deficientes, provocar a chamada língua negra em cães, somente pecando por achar que a deficiência seria de aminoácidos. Mais tarde foi sabido que a pelagra poderia ser prevenida com o uso de preparações de vitamina B solúvel na água contendo um fator resistente ao calor. Lá por 1935 foi que Warburg isolou a amida do ácido nicotínico (nicotinamida) de coenzimas das hemácias do cavalo. Dois anos após, Elvehjem mostrou que a nicotinamida contida no fígado era eficaz no tratamento da pelagra e da língua negra. Goldberger, com grande perspicácia, mostrou que o triptofano podia curar a pelagra humana por ser convertido pelo organismo em ácido nicotínico.
A pá de cal no assunto foi colocada por Goldshmidt, já em 1958, quando provocou pelagra experimentalmente em humanos alimentados com dieta pobre em ácido nicotínico e triptofano. Como pode ser sintetizada a partir do aminoácido essencial triptofano (cada 60 mg de triptofano fornecido pela dieta dá origem a 1 mg de niacina), alguns autores não consideram a niacina uma verdadeira vitamina.
A pelagra já foi endêmica na América do Sul e outras partes do mundo, o que, era determinado pelo alto consumo do milho. Mas, nem tudo está bem explicado porque há povos que consomem muito milho como principal fonte proteica e não têm pelagra. E, embora o milho tenha pouca niacina, há outros cereais com conteúdo semelhante que não são associados à pelagra. A doença seria mais por um balanceamento inadequado de aminoácidos na dieta ou um problema de complexidade muito maior? Mistério, diria o nosso pároco numa introspectiva homilia. A moagem do milho prejudica a bioavaliabilidade da niacina.
O ácido nicotínico somente exerce as suas funções orgânicas ao ser convertido em nicotinamida adenina dinucleotídeo (NAD) ou nicotinamida adenina dinucleotídeo fosfato (NADP), que servem como coenzima para uma série de proteínas que catalizam uma série de reações de oxi-redução essenciais para a respiração dos tecidos. O termo niacina engloba o ácido nicotínico, a NAD e a NADP.
Os sinais de intoxicação pelo ácido nicotínico (ácido piridino 3 carboxílico) são o prurido (coceira), o rubor da pele, intoxicação hepática, ativação de úlcera péptica e alterações gastrintestinais.
O sinais da dermatose simétrica provocada pela pelagra lembram os da queimadura solar e surgem no dorso da mão (pode evoluir como lesão em luva, a luva pelagrosa), testa, pescoço (colar de Casal)e pés, muitas vezes como uma bota (bota pelagrosa); como vê-se, aparece nas regiões expostas aos raios solares e parece ser devida a fotossensibilização (para outros autores, os raios solares seriam apenas fatores irritantes que piorariam a dermatose) e termina em descamação e formação de cicatrizes (algumas vezes predominam as bolhas e as vesículas, o tipo úmido).
Apresenta a pelagra também sinais digestivos, como estomatite, glossite (primeiramente aparecem as lesões vermelhas e inchadas na ponta e nas margens laterais, evoluindo para atingir toda a língua. Podem haver ulcerações), muitas vezes associadas a vaginite, enterite e diarréia, náuseas e vômitos, e sinais neurológicos como insônia, depressão, fadiga, apatia, perda da memória, desorientação, halucinações, demência, além de distúrbios sensoriais e motores dos nervos periféricos (acha-se que a polineurite periférica seja devida à ausência de outras vitaminas). É a síndrome dos três D (diarréia, demência, dermatite). Os sinais mentais talvez surjam pela queda na conversão do triptofano em serotonina, o chamado hormônio do prazer.
As fontes principais do ácido nicotínico são as carnes de gado, a carne magra de porco, o fígado, o frango, o peixe (o salmão é muito rico), os grãos integrais, os cereais enriquecidos, os legumes e as nozes. O triptofano, aminoácido essencial, é fornecido principalmente pelas proteínas animais. O leite e o ovo contêm pouca niacina, mas são ricos em triptofano, o que, lhes confere papel importante na profilaxia da pelagra. Nunca é demais lembrar que as proteínas do leite e do ovo são as únicas da dieta comum que são completas, isso é, possuem todos os aminoácidos essenciais. Parte da niacina contida em alguns alimentos, como certos cereais, está na forma conjugada e não pode ser disponibilizada do ponto de vista nutricional. Por ser estável, os níveis de niacina pouco são afetados pelo cozimento ou a fervura dos alimentos.
Tanto o ácido nicotínico como a nicotinamida são absorvidos por todo o trato intestinal, por vias passiva e ativa, numa média entre 3 a 4g/dia num adulto humano, e distribuem-se por todos os tecidos. Um quinto é transformado em ácido nicotinúrico e o restante é eliminado na urina principalmente como carboxamida metil piridona e metilnicotinamida.
As necessidades humanas diárias variam de 5 mg/dia na infância até 20 mg/dia nas mulheres em lactação. Ao contrário de outras vitaminas, as necessidades parecem não aumentar durante a gravidez. As necessidades aumentam em quadros em que o triptofano é altamente catabolizado, como a síndrome carcinóide, ou pobremente absorvido como na doença de Hartnup (herdada, autossômica recessiva, com deficiência do transporte de aminoácidos e que, entre os sinais clínicos, apresenta dermatose semelhante à pelagra). Lembrar que altas doses de niacina (10 vezes maior do que as doses recomendadas nos pássaros) podem provocar flushing, sensação de calor e de queimação e, nos casos mais graves, evoluir para hepatoxiidade, colestase (retenção biliar) e icterícia (amarelão da pele e mucosas determinado pela bilirrubina).
Um dado terapêutico interessante: embora a pelagra responda bem ao ácido nicotínico ou seus congêneres, a neurite periférica não responde a eles e sim à tiamina. Alguns autores aconselham acrescentar a riboflavina e a piridoxina nos casos de pelagra mais resistente. A evolução da pelagra é longa e progressiva, às vezes durante anos, e a morte acontece normalmente por complicações secundárias. Volto a lembrar que a deficiência geralmente não se restringe a uma só vitamina do complexo B, o que, determina que um grupo delas deve sempre entrar no raciocínio de quem traça um esquema profilático e terapêutico."
By: Ivana Melo
Fonte: http://www.cobrap.org.br/site/artigos_vis.php?id=414
Bom, acima tem uma explicação bem simples sobre a niacina e a pelagra, principais aspectos e algumas curiosidades. Obs.: "Os sinais mentais talvez surjam pela queda na conversão do triptofano em serotonina", creio que não há relação com demência e sim a a síndrome de carcinóide.
Goldberger, outro grande estudioso das vitaminas conseguiu, usando dietas deficientes, provocar a chamada língua negra em cães, somente pecando por achar que a deficiência seria de aminoácidos. Mais tarde foi sabido que a pelagra poderia ser prevenida com o uso de preparações de vitamina B solúvel na água contendo um fator resistente ao calor. Lá por 1935 foi que Warburg isolou a amida do ácido nicotínico (nicotinamida) de coenzimas das hemácias do cavalo. Dois anos após, Elvehjem mostrou que a nicotinamida contida no fígado era eficaz no tratamento da pelagra e da língua negra. Goldberger, com grande perspicácia, mostrou que o triptofano podia curar a pelagra humana por ser convertido pelo organismo em ácido nicotínico.
A pá de cal no assunto foi colocada por Goldshmidt, já em 1958, quando provocou pelagra experimentalmente em humanos alimentados com dieta pobre em ácido nicotínico e triptofano. Como pode ser sintetizada a partir do aminoácido essencial triptofano (cada 60 mg de triptofano fornecido pela dieta dá origem a 1 mg de niacina), alguns autores não consideram a niacina uma verdadeira vitamina.
A pelagra já foi endêmica na América do Sul e outras partes do mundo, o que, era determinado pelo alto consumo do milho. Mas, nem tudo está bem explicado porque há povos que consomem muito milho como principal fonte proteica e não têm pelagra. E, embora o milho tenha pouca niacina, há outros cereais com conteúdo semelhante que não são associados à pelagra. A doença seria mais por um balanceamento inadequado de aminoácidos na dieta ou um problema de complexidade muito maior? Mistério, diria o nosso pároco numa introspectiva homilia. A moagem do milho prejudica a bioavaliabilidade da niacina.
O ácido nicotínico somente exerce as suas funções orgânicas ao ser convertido em nicotinamida adenina dinucleotídeo (NAD) ou nicotinamida adenina dinucleotídeo fosfato (NADP), que servem como coenzima para uma série de proteínas que catalizam uma série de reações de oxi-redução essenciais para a respiração dos tecidos. O termo niacina engloba o ácido nicotínico, a NAD e a NADP.
Os sinais de intoxicação pelo ácido nicotínico (ácido piridino 3 carboxílico) são o prurido (coceira), o rubor da pele, intoxicação hepática, ativação de úlcera péptica e alterações gastrintestinais.
O sinais da dermatose simétrica provocada pela pelagra lembram os da queimadura solar e surgem no dorso da mão (pode evoluir como lesão em luva, a luva pelagrosa), testa, pescoço (colar de Casal)e pés, muitas vezes como uma bota (bota pelagrosa); como vê-se, aparece nas regiões expostas aos raios solares e parece ser devida a fotossensibilização (para outros autores, os raios solares seriam apenas fatores irritantes que piorariam a dermatose) e termina em descamação e formação de cicatrizes (algumas vezes predominam as bolhas e as vesículas, o tipo úmido).
Apresenta a pelagra também sinais digestivos, como estomatite, glossite (primeiramente aparecem as lesões vermelhas e inchadas na ponta e nas margens laterais, evoluindo para atingir toda a língua. Podem haver ulcerações), muitas vezes associadas a vaginite, enterite e diarréia, náuseas e vômitos, e sinais neurológicos como insônia, depressão, fadiga, apatia, perda da memória, desorientação, halucinações, demência, além de distúrbios sensoriais e motores dos nervos periféricos (acha-se que a polineurite periférica seja devida à ausência de outras vitaminas). É a síndrome dos três D (diarréia, demência, dermatite). Os sinais mentais talvez surjam pela queda na conversão do triptofano em serotonina, o chamado hormônio do prazer.
As fontes principais do ácido nicotínico são as carnes de gado, a carne magra de porco, o fígado, o frango, o peixe (o salmão é muito rico), os grãos integrais, os cereais enriquecidos, os legumes e as nozes. O triptofano, aminoácido essencial, é fornecido principalmente pelas proteínas animais. O leite e o ovo contêm pouca niacina, mas são ricos em triptofano, o que, lhes confere papel importante na profilaxia da pelagra. Nunca é demais lembrar que as proteínas do leite e do ovo são as únicas da dieta comum que são completas, isso é, possuem todos os aminoácidos essenciais. Parte da niacina contida em alguns alimentos, como certos cereais, está na forma conjugada e não pode ser disponibilizada do ponto de vista nutricional. Por ser estável, os níveis de niacina pouco são afetados pelo cozimento ou a fervura dos alimentos.
Tanto o ácido nicotínico como a nicotinamida são absorvidos por todo o trato intestinal, por vias passiva e ativa, numa média entre 3 a 4g/dia num adulto humano, e distribuem-se por todos os tecidos. Um quinto é transformado em ácido nicotinúrico e o restante é eliminado na urina principalmente como carboxamida metil piridona e metilnicotinamida.
As necessidades humanas diárias variam de 5 mg/dia na infância até 20 mg/dia nas mulheres em lactação. Ao contrário de outras vitaminas, as necessidades parecem não aumentar durante a gravidez. As necessidades aumentam em quadros em que o triptofano é altamente catabolizado, como a síndrome carcinóide, ou pobremente absorvido como na doença de Hartnup (herdada, autossômica recessiva, com deficiência do transporte de aminoácidos e que, entre os sinais clínicos, apresenta dermatose semelhante à pelagra). Lembrar que altas doses de niacina (10 vezes maior do que as doses recomendadas nos pássaros) podem provocar flushing, sensação de calor e de queimação e, nos casos mais graves, evoluir para hepatoxiidade, colestase (retenção biliar) e icterícia (amarelão da pele e mucosas determinado pela bilirrubina).
Um dado terapêutico interessante: embora a pelagra responda bem ao ácido nicotínico ou seus congêneres, a neurite periférica não responde a eles e sim à tiamina. Alguns autores aconselham acrescentar a riboflavina e a piridoxina nos casos de pelagra mais resistente. A evolução da pelagra é longa e progressiva, às vezes durante anos, e a morte acontece normalmente por complicações secundárias. Volto a lembrar que a deficiência geralmente não se restringe a uma só vitamina do complexo B, o que, determina que um grupo delas deve sempre entrar no raciocínio de quem traça um esquema profilático e terapêutico."
By: Ivana Melo
Fonte: http://www.cobrap.org.br/site/artigos_vis.php?id=414
Bom, acima tem uma explicação bem simples sobre a niacina e a pelagra, principais aspectos e algumas curiosidades. Obs.: "Os sinais mentais talvez surjam pela queda na conversão do triptofano em serotonina", creio que não há relação com demência e sim a a síndrome de carcinóide.
Sintomas da Pelagra....

Diarréia.
Fonte: http://www.overmundo.com.br/_banco/multiplas/12103528211_diarreia.jpg
 Demência.
Demência.Fonte: http://www.apuntesdepsicologia.com/img/demencia-semantica1.jpg

Dermatite
Fonte: http://www.uftm.edu.br/instpub/fmtm/nutrologia/Pelagra%20cicatrizando.jpg
Os "3D" da pelagra.
By: Ivana Melo
Pelagra x Síndrome carcinóide
O artigo a seguir aborda sobre a síndrome carcinóide, mas para fins didáticos não analisaremos o artigo por completo e sim a parte histórica.
GASTRO-INTESTINAL CARCINOIDS are slow growing neoplasms as compared with adenocarcinomas, but they can also behave aggressively. They are derived from neoplastic proliferation of enterochromaffin (ECL) or Kulchitsky cells (1).
In 1888, Lubarsch first described a patient with multiple carcinoids of the ileum but regarded them as carcinomas (2). Two years later, Ransom first described the classical symptomatology of the carcinoid syndrome in a patient with an ileal carcinoid tumor and hepatic metastasis (3). However, it was Oberndorfer in 1907, who coined the term "karzinoide" to describe these tumors, which he believed to behave in a more benign fashion than adenocarcinomas (4). In 1963, Williams and Sandler classified carcinoids according to their embryologic site of origin as foregut carcinoids (respiratory tract, stomach, duodenum, biliary system, and pancreas), midgut carcinoids (small intestine, appendix, cecum, and proximal colon), and hindgut carcinoids (distal colon and rectum) (5). However, these lesions exhibit a high degree of morphologic and biologic heterogeneity and a more generic term, neuroendocrine tumor (NET) has been introduced to replace the term carcinoid. Such lesions are currently referred to as gastroenteropancreatic (GEP) NETs (GEP-NETs). According to the WHO classification, distinction was made between well-differentiated NETs (benign behavior or uncertain malignant potential), well-differentiated neuroendocrine carcinomas (low-grade malignancy), and poorly differentiated (usually small cell) neuroendocrine carcinomas of high-grade malignancy. Nevertheless, the term carcinoid was not abandoned and for GEP-NETs, it is used synonymously with the term "well-differentiated NET". The term "malignant carcinoid" is used synonymously with the term well-differentiated neuroendocrine carcinoma (6). The differentiation is based on tumor morphology, tumor size (in general larger tumors are more aggressive), and the presence or absence of local invasion and/or metastasis, thus reflecting biological behavior. Most NETs are well-differentiated tumors that are characterized by a solid trabecular or glandular structure, tumor cell monomorphism with absent or low cytological atypia, and a low mitotic (<> 10 mitoses/mm2) and proliferative status (> 15% Ki-67 positive cells), diffuse reactivity for cytosolic markers, and scant or weak reactivity for granular markers or neurosecretory products (1).
Carcinoid lesions are the most common NETs and compose approximately 50% of all NETs of the gastrointestinal tract. In most instances, they are discovered incidentally at the time of surgery for other abdominal disorders. Their presence may be undetectable for years without obvious signs or symptoms. Evidence for this observation is supported by their relatively high incidence in large autopsy series (7). When symptoms do occur, they are due either to local tumor mass effects, the effects of tumor-engendered fibrosis, or to the secreted bioactive products from the neoplasm. Symptoms caused by local tumor effects include vague abdominal pain (invasion, intussusception, fibrous adhesions, hypermotility), which is often undiagnosed or leads to erroneous diagnoses like irritable bowel syndrome (8,9).
Carcinoids have protean clinical presentations, depending on what combination of bioactive substances is secreted. One of their main characteristics of the enterochromaffin (ECL) or Kulchitsky cells is the synthesis, storage, and secretion of serotonin. Serotonin (5-hydroxytrypamine, 5-HT) is synthesized from tryptophan through its precursor, 5-hydroxy tryptophan (5-HTP), and subsequently metabolized to 5-hydroxyindoleacetic acid (5-HIAA), which is excreted in the urine. In addition to serotonin, carcinoid tumors may also secrete other hormones such as corticotrophin (ACTH), histamine, dopamine, substance P, neurotensin, prostaglandins, kallikrein, and tachykinins. In normal subjects, approximately 99% of tryptophan is used for the synthesis of nicotinic acid (niacin), and 1% or less is converted to 5-HT. In patients with carcinoid tumors, there is a shift toward the production of 5-HT and eventually 5-HIAA. This may lead to tryptophan deficiency and pellagra might ensue as a result of nicotinic acid deficiency (9). When 5-HT and other products are secreted into the portal circulation, they are efficiently metabolized by the liver and do not usually cause any systemic signs or symptoms. However, when liver metastases are present or when the primary lesions are found in the bronchus and/or ovaries, the systemic features of the carcinoid syndrome become more evident. This classical syndrome occurs in fewer than 10% of patients, and its most typical clinical manifestations include cutaneous flushing most commonly of the face, neck, and upper chest and diarrhea, occurring in up to 75%. Less frequent manifestations include cardiac valvular abnormalities (plaque-like, fibrous endocardial thickening that principally involves the right side of the heart (causing tricuspid regurgitation tricuspid stenosis, pulmonary regurgitation, and pulmonary stenosis), bronchoconstriction and (as already mentioned) pellagra. Foregut carcinoids can secrete 5-HTP, histamine and polypeptide hormones like ACTH. The can produce a characteristic clinical syndrome known as "atypical" carcinoid syndrome. Midgut carcinoids release 5-HT and other vasoactive compounds such as kinins, prostaglandins, and substance P and they are more likely to cause the classic carcinoid syndrome with the development of hepatic metastases. Hindgut carcinoid tumors rarely contain 5-HT and usually do not present with the carcinoid syndrome; however. The symptoms of the carcinoid syndrome can be both of variable intensity as well as paroxysmal, responding intermittently to a particular "trigger" agent, such as alcohol, cheese, coffee (these are serotonin-rich foods), or exercise (8,10,11).
Many carcinoid tumors exhibit a significant association with other non-carcinoid tumors of various histological types. A relatively large percentage of carcinoids are multicentric.
Because carcinoid tumors frequently present with obscure clinical manifestations, numerous investigatory procedures are often undertaken prior to establishing the correct diagnosis. Although clinical diagnosis is based on symptoms, biochemical confirmation is necessary. The diagnostic strategies employed usually depend on the individual clinical presentation (8,10,11)."
Fonte Bibliográfica: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27302005000500028&lng=pt&nrm=isso
Com base no artigo, percebe-se a influência da síndrome com a pelagra, pois a secreção liberada pelo tumor, no caso a enterochromaffin (ECL), está associada com a produção de serotonina. Para que ocorra síntese de serotonina, deve-se ter triptofano como substrato, logo o triptofano presente no organismo ao invés de ser convertido a niacina - vitamina anti-pelagra- em grande escala é convertido em serotonina, diminuindo os níveis de niacina provocando a pelagra.
By: Ivana Melo
" HISTORICAL OVERVIEW
GASTRO-INTESTINAL CARCINOIDS are slow growing neoplasms as compared with adenocarcinomas, but they can also behave aggressively. They are derived from neoplastic proliferation of enterochromaffin (ECL) or Kulchitsky cells (1).
In 1888, Lubarsch first described a patient with multiple carcinoids of the ileum but regarded them as carcinomas (2). Two years later, Ransom first described the classical symptomatology of the carcinoid syndrome in a patient with an ileal carcinoid tumor and hepatic metastasis (3). However, it was Oberndorfer in 1907, who coined the term "karzinoide" to describe these tumors, which he believed to behave in a more benign fashion than adenocarcinomas (4). In 1963, Williams and Sandler classified carcinoids according to their embryologic site of origin as foregut carcinoids (respiratory tract, stomach, duodenum, biliary system, and pancreas), midgut carcinoids (small intestine, appendix, cecum, and proximal colon), and hindgut carcinoids (distal colon and rectum) (5). However, these lesions exhibit a high degree of morphologic and biologic heterogeneity and a more generic term, neuroendocrine tumor (NET) has been introduced to replace the term carcinoid. Such lesions are currently referred to as gastroenteropancreatic (GEP) NETs (GEP-NETs). According to the WHO classification, distinction was made between well-differentiated NETs (benign behavior or uncertain malignant potential), well-differentiated neuroendocrine carcinomas (low-grade malignancy), and poorly differentiated (usually small cell) neuroendocrine carcinomas of high-grade malignancy. Nevertheless, the term carcinoid was not abandoned and for GEP-NETs, it is used synonymously with the term "well-differentiated NET". The term "malignant carcinoid" is used synonymously with the term well-differentiated neuroendocrine carcinoma (6). The differentiation is based on tumor morphology, tumor size (in general larger tumors are more aggressive), and the presence or absence of local invasion and/or metastasis, thus reflecting biological behavior. Most NETs are well-differentiated tumors that are characterized by a solid trabecular or glandular structure, tumor cell monomorphism with absent or low cytological atypia, and a low mitotic (<> 10 mitoses/mm2) and proliferative status (> 15% Ki-67 positive cells), diffuse reactivity for cytosolic markers, and scant or weak reactivity for granular markers or neurosecretory products (1).
Carcinoid lesions are the most common NETs and compose approximately 50% of all NETs of the gastrointestinal tract. In most instances, they are discovered incidentally at the time of surgery for other abdominal disorders. Their presence may be undetectable for years without obvious signs or symptoms. Evidence for this observation is supported by their relatively high incidence in large autopsy series (7). When symptoms do occur, they are due either to local tumor mass effects, the effects of tumor-engendered fibrosis, or to the secreted bioactive products from the neoplasm. Symptoms caused by local tumor effects include vague abdominal pain (invasion, intussusception, fibrous adhesions, hypermotility), which is often undiagnosed or leads to erroneous diagnoses like irritable bowel syndrome (8,9).
Carcinoids have protean clinical presentations, depending on what combination of bioactive substances is secreted. One of their main characteristics of the enterochromaffin (ECL) or Kulchitsky cells is the synthesis, storage, and secretion of serotonin. Serotonin (5-hydroxytrypamine, 5-HT) is synthesized from tryptophan through its precursor, 5-hydroxy tryptophan (5-HTP), and subsequently metabolized to 5-hydroxyindoleacetic acid (5-HIAA), which is excreted in the urine. In addition to serotonin, carcinoid tumors may also secrete other hormones such as corticotrophin (ACTH), histamine, dopamine, substance P, neurotensin, prostaglandins, kallikrein, and tachykinins. In normal subjects, approximately 99% of tryptophan is used for the synthesis of nicotinic acid (niacin), and 1% or less is converted to 5-HT. In patients with carcinoid tumors, there is a shift toward the production of 5-HT and eventually 5-HIAA. This may lead to tryptophan deficiency and pellagra might ensue as a result of nicotinic acid deficiency (9). When 5-HT and other products are secreted into the portal circulation, they are efficiently metabolized by the liver and do not usually cause any systemic signs or symptoms. However, when liver metastases are present or when the primary lesions are found in the bronchus and/or ovaries, the systemic features of the carcinoid syndrome become more evident. This classical syndrome occurs in fewer than 10% of patients, and its most typical clinical manifestations include cutaneous flushing most commonly of the face, neck, and upper chest and diarrhea, occurring in up to 75%. Less frequent manifestations include cardiac valvular abnormalities (plaque-like, fibrous endocardial thickening that principally involves the right side of the heart (causing tricuspid regurgitation tricuspid stenosis, pulmonary regurgitation, and pulmonary stenosis), bronchoconstriction and (as already mentioned) pellagra. Foregut carcinoids can secrete 5-HTP, histamine and polypeptide hormones like ACTH. The can produce a characteristic clinical syndrome known as "atypical" carcinoid syndrome. Midgut carcinoids release 5-HT and other vasoactive compounds such as kinins, prostaglandins, and substance P and they are more likely to cause the classic carcinoid syndrome with the development of hepatic metastases. Hindgut carcinoid tumors rarely contain 5-HT and usually do not present with the carcinoid syndrome; however. The symptoms of the carcinoid syndrome can be both of variable intensity as well as paroxysmal, responding intermittently to a particular "trigger" agent, such as alcohol, cheese, coffee (these are serotonin-rich foods), or exercise (8,10,11).
Many carcinoid tumors exhibit a significant association with other non-carcinoid tumors of various histological types. A relatively large percentage of carcinoids are multicentric.
Because carcinoid tumors frequently present with obscure clinical manifestations, numerous investigatory procedures are often undertaken prior to establishing the correct diagnosis. Although clinical diagnosis is based on symptoms, biochemical confirmation is necessary. The diagnostic strategies employed usually depend on the individual clinical presentation (8,10,11)."
Fonte Bibliográfica: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27302005000500028&lng=pt&nrm=isso
Com base no artigo, percebe-se a influência da síndrome com a pelagra, pois a secreção liberada pelo tumor, no caso a enterochromaffin (ECL), está associada com a produção de serotonina. Para que ocorra síntese de serotonina, deve-se ter triptofano como substrato, logo o triptofano presente no organismo ao invés de ser convertido a niacina - vitamina anti-pelagra- em grande escala é convertido em serotonina, diminuindo os níveis de niacina provocando a pelagra.
By: Ivana Melo
"Pelagra Endógena" - Doença de Hartnup.... ???
" Pelagra endógena e ataxia cerebelar sem aminoacidúria. Doença de Hartnup?
RESUMO
Menino, 7 anos, com história de convulsão, hiperpigmentação cutânea em áreas de exposição solar e episódios recorrentes de ataxia cerebelar. Estabelecido diagnóstico clínico de doença de Hartnup, foi tratado com nicotinamida, com melhora. Análises não confirmaram aminoacidúria ou outras alterações metabólicas. Na doença de Hartnup ocorre defeito no transporte renal e intestinal de aminoácidos neutros, reduzindo triptofano disponível para produção de niacina. Cursa com ataxia cerebelar intermitente, erupções cutâneas pelagróides e distúrbios mentais. Aminoacidúria em cromatografia urinária confirma diagnóstico, porém são descritos casos compatíveis com doença de Hartnup sem aminoacidúria.
Palavras-chave: Aminoacidúria renal; Doença de Hartnup; Pelagra
INTRODUÇÃO
A doença de Hartnup é condição genética autossômica recessiva rara, que acomete principalmente crianças entre cinco e 15 anos de idade,1 descrita por Baron e cols.2 em 1956. Sua fisiopatologia está relacionada a defeito no transporte tubular proximal renal e intestinal jejunal de aminoácidos neutros, sendo a gênese de suas manifestações clínicas atribuída à queda dos níveis de niacina (vitamina B3), ocasionada pela diminuição da absorção de seu precursor, o triptofano.
Múltiplas formas de apresentação podem ser reconhecidas, desde indivíduos totalmente assintomáticos, passando pela forma mais freqüente identificada por dermatite fotossensível do tipo pelagróide associada a ataxia cerebelar intermitente e sintomas neuropsíquicos, podendo até mesmo em alguns casos desenvolver quadros mais graves com lesões neurodegenerativas progressivas e morte. Exposição à luz solar, febre, uso de sulfonamidas, estresse emocional, infecções intercorrentes e dieta irregular ou inadequada são fatores descritos como possíveis desencadeantes dos sinais e sintomas.
O diagnóstico da doença de Hartnup é estabelecido pela identificação de hiperaminoacidúria na cromatografia urinária. A ausência de hiperaminoacidúria em pacientes com quadro clínico de doença de Hartnup vem sendo descrita por alguns autores. O tratamento baseia-se na administração oral de nicotinamida (40 a 250mg/dia).
RELATO DO CASO
Menino de sete anos, branco, procedente de zona rural, filho único de pais sadios não consangüíneos, foi assistido em serviço de pronto atendimento em julho de 2003, ocasião na qual a mãe relatou história de crise convulsiva generalizada, de início súbito, no domicílio, uma hora antes. Negou passado recente ou remoto de trauma crânio-encefálico, infecções do sistema nervoso central, crises semelhantes ou qualquer outra doença associada nem uso rotineiro de qualquer medicação.
Mãe refere que em duas ocasiões (junho de 2001 e julho de 2002) a criança desenvolveu lesões cutâneas eritêmato-descamativas, não pruriginosas, inicialmente vinhosas que evoluíam para áreas avermelhadas com descamação, sempre restritas a regiões de exposição ao sol, associadas a alteração importante da marcha (cambaleante) que perduravam ao longo de duas ou três semanas com resolução espontânea e concomitante das lesões cutâneas e da marcha, recebendo, na ocasião, o diagnóstico de pelagra.
Na admissão hospitalar chamava a atenção aspecto ressecado da pele, eritematosa com áreas de descamação superficial localizadas em face, nuca, colo, dorso de antebraços, mãos ,além de pernas e pés.Negava alteração do hábito intestinal. Neurologicamente apresentava-se consciente, auto e alodesorientado, hiperreflexia global de tendões profundos, sem sinais de irritação meníngea, marcha atáxica (base alargada e articulações enrijecidas) que evoluiu atingindo a inabilidade para deambulação. Exame de fundo de olho normal. Demais sistemas sem alterações. Avaliação antropométrica: 25kg de peso (p75%), 118cm (p25%) de estatura.
Por ocasião da internação foram realizados hemograma, EAS e urina de 24 horas, glicemia, dosagem de eletrólitos e proteínas (total e frações), provas de função hepática e renal, análise do LCR (incluindo exame bacterioscópico, pesquisa de fungos, toxoplasmose e VDRL), radiografia de tórax e crânio, sendo considerados todos os resultados normais. A tomografia craniana computadorizada e a ressonâncea magnética do crânio não revelaram anomalias. Eletroencefalograma (EEG) de repouso e ativado pela hiperpnéia evidenciou desorganização difusa do traçado. Sua idade óssea foi calculada em sete anos, segundo critérios de Pyle para sexo masculino.
A cromatografia plana e bidimensional de aminoácidos em papel não revelou elevação na excreção urinária de aminoácidos. Níveis séricos de aminoácidos mostraram-se dentro dos padrões de normalidade. Não foi detectado indican urinário, tampouco qualquer outra alteração durante realização de exames de rastreamento para erros inatos do metabolismo. Não há na família relato de quadro clínico semelhante ou distúrbios sugestivos de doença metabólica específica.
Paciente permaneceu internado durante 11 dias em uso de complexo polivitamínico B (80mg/dia de nicotinamida). Com melhora progressiva do quadro cutâneo e neurológico a partir do quarto dia de internação, recebeu alta com marcha atípica e resolução quase completa da dermatite pelagróide. Nos três meses subseqüentes retornou ao ambulatório de pediatria, assintomático e em uso de complexo polivitamínico B (20mg/dia de nicotinamida).
DISCUSSÃO
As manifestações clínicas de dermatite pelagróide – caracterizada por lesões eritêmato-descamativas em áreas de exposição ao sol – associadas ao surgimento concomitante de ataxia cerebelar intermitente, atingindo a inabilidade para deambulação, enquadram perfeitamente o paciente descrito ao diagnóstico clínico de doença de Hartnup em sua apresentação clássica. Além disso, achados secundários, tais como labilidade emocional, hiper-reflexia global de tendões profundos, crise convulsiva e alterações difusas no EEG, foram citados por outros autores como possíveis achados em pacientes com doença de Hartnup.
O caráter intermitente das alterações clínicas com fases de exacerbação verificadas principalmente no período de junho-julho, idêntico ao relatado por Da Gloria e cols., muito provavelmente, segundo relato da mãe, deve-se, nesse caso, à significativa exposição do paciente à luz solar nesse período, coincidente com o de férias escolares, ao que se soma o fato de as estações do ano serem pouco definidas na região.
Goulon e cols.5 em seu trabalho subdividem os casos de pelagra em exógenos e endógenos. A pelagra exógena está relacionada à hipovitaminose B3 induzida principalmente por dieta pobre em triptofano e não está associada a aumento da excreção renal de aminoácidos, ao contrário da pelagra endógena, cujo principal exemplo é a doença de Hartnup, sendo a queda dos níveis de niacina resultado de defeito na absorção renal e intestinal do triptofano.
Pelagra carencial deve ser considerada o principal diagnóstico diferencial, afastado, no entanto, pela ausência dos distúrbios gastrointestinais típicos da síndrome (em especial a diarréia) e pela diferença entre as manifestações neuropsíquicas apresentadas pelo paciente e a demência característica da pelagra exógena. Além disso, a ingesta diária de leite de vaca (alimento rico em triptofano), a sazonalidade das alterações clínicas e a não-observância de sinais e sintomas semelhantes em outras crianças da comunidade rural habitada pelo paciente são também fatores que se somam para afastar o diagnóstico de pelagra nutricional. Outras etiologias possíveis para a pelagra endógena, além da doença de Hartnup, seriam o uso prolongado de isoniazida, de 6-mercaptopurina e tumores carcinóides, situações essas não verificadas no paciente em questão.5
Assim, frente ao quadro clínico apresentado pelo paciente, somado à eficácia do tratamento proposto, responsável pela remissão tanto de lesões cutâneas como das alterações neurológicas, sugere-se diagnóstico de pelagra endógena. Porém, tal como nos casos descritos por Goulon e cols. (França), Da Gloria e cols.(Brasil), Borrie e Lewis e Tada e cols. (Japão), neste também não se evidenciou hiperaminoacidúria típica da doença de Hartnup quando da realização da cromatografia plana e bidimensional de aminoácidos em papel.
Possível explicação já aventada para a não-verificação de aminoacidúria em pacientes com quadro clínico da doença de Hartnup seria uma alteração no metabolismo do triptofano que acarretaria distúrbio em sua transformação em cirunenina por defeito da enzima triptofano-pirrolase. Ou, ainda, como no caso descrito por Borrie e Lewis,a dicotomia entre quadro clínico e achados laboratoriais poderia ser devida a possível genótipo heterozigoto do paciente avaliado.
Entretanto, assim como os casos apresentados por Goulon e cols. e Da Gloria e cols., não foi possível precisar se o déficit de niacina seria resultado de alteração no metabolismo ou transporte renal/intestinal do triptofano. Da Gloria e cols. questionam se a presença de sinais e sintomas da doença de Hartnup sem associação com qualquer disfunção metabólica até agora identificada ou com níveis aumentados de aminoácidos na urina poderia até mesmo tratar-se de uma nova entidade, o que, acreditam os autores, deve ser alvo de investigação, dadas a singularidade e a semelhança do caso aqui descrito com os demais citados."
RESUMO
Menino, 7 anos, com história de convulsão, hiperpigmentação cutânea em áreas de exposição solar e episódios recorrentes de ataxia cerebelar. Estabelecido diagnóstico clínico de doença de Hartnup, foi tratado com nicotinamida, com melhora. Análises não confirmaram aminoacidúria ou outras alterações metabólicas. Na doença de Hartnup ocorre defeito no transporte renal e intestinal de aminoácidos neutros, reduzindo triptofano disponível para produção de niacina. Cursa com ataxia cerebelar intermitente, erupções cutâneas pelagróides e distúrbios mentais. Aminoacidúria em cromatografia urinária confirma diagnóstico, porém são descritos casos compatíveis com doença de Hartnup sem aminoacidúria.
Palavras-chave: Aminoacidúria renal; Doença de Hartnup; Pelagra
INTRODUÇÃO
A doença de Hartnup é condição genética autossômica recessiva rara, que acomete principalmente crianças entre cinco e 15 anos de idade,1 descrita por Baron e cols.2 em 1956. Sua fisiopatologia está relacionada a defeito no transporte tubular proximal renal e intestinal jejunal de aminoácidos neutros, sendo a gênese de suas manifestações clínicas atribuída à queda dos níveis de niacina (vitamina B3), ocasionada pela diminuição da absorção de seu precursor, o triptofano.
Múltiplas formas de apresentação podem ser reconhecidas, desde indivíduos totalmente assintomáticos, passando pela forma mais freqüente identificada por dermatite fotossensível do tipo pelagróide associada a ataxia cerebelar intermitente e sintomas neuropsíquicos, podendo até mesmo em alguns casos desenvolver quadros mais graves com lesões neurodegenerativas progressivas e morte. Exposição à luz solar, febre, uso de sulfonamidas, estresse emocional, infecções intercorrentes e dieta irregular ou inadequada são fatores descritos como possíveis desencadeantes dos sinais e sintomas.
O diagnóstico da doença de Hartnup é estabelecido pela identificação de hiperaminoacidúria na cromatografia urinária. A ausência de hiperaminoacidúria em pacientes com quadro clínico de doença de Hartnup vem sendo descrita por alguns autores. O tratamento baseia-se na administração oral de nicotinamida (40 a 250mg/dia).
RELATO DO CASO
Menino de sete anos, branco, procedente de zona rural, filho único de pais sadios não consangüíneos, foi assistido em serviço de pronto atendimento em julho de 2003, ocasião na qual a mãe relatou história de crise convulsiva generalizada, de início súbito, no domicílio, uma hora antes. Negou passado recente ou remoto de trauma crânio-encefálico, infecções do sistema nervoso central, crises semelhantes ou qualquer outra doença associada nem uso rotineiro de qualquer medicação.
Mãe refere que em duas ocasiões (junho de 2001 e julho de 2002) a criança desenvolveu lesões cutâneas eritêmato-descamativas, não pruriginosas, inicialmente vinhosas que evoluíam para áreas avermelhadas com descamação, sempre restritas a regiões de exposição ao sol, associadas a alteração importante da marcha (cambaleante) que perduravam ao longo de duas ou três semanas com resolução espontânea e concomitante das lesões cutâneas e da marcha, recebendo, na ocasião, o diagnóstico de pelagra.
Na admissão hospitalar chamava a atenção aspecto ressecado da pele, eritematosa com áreas de descamação superficial localizadas em face, nuca, colo, dorso de antebraços, mãos ,além de pernas e pés.Negava alteração do hábito intestinal. Neurologicamente apresentava-se consciente, auto e alodesorientado, hiperreflexia global de tendões profundos, sem sinais de irritação meníngea, marcha atáxica (base alargada e articulações enrijecidas) que evoluiu atingindo a inabilidade para deambulação. Exame de fundo de olho normal. Demais sistemas sem alterações. Avaliação antropométrica: 25kg de peso (p75%), 118cm (p25%) de estatura.
Por ocasião da internação foram realizados hemograma, EAS e urina de 24 horas, glicemia, dosagem de eletrólitos e proteínas (total e frações), provas de função hepática e renal, análise do LCR (incluindo exame bacterioscópico, pesquisa de fungos, toxoplasmose e VDRL), radiografia de tórax e crânio, sendo considerados todos os resultados normais. A tomografia craniana computadorizada e a ressonâncea magnética do crânio não revelaram anomalias. Eletroencefalograma (EEG) de repouso e ativado pela hiperpnéia evidenciou desorganização difusa do traçado. Sua idade óssea foi calculada em sete anos, segundo critérios de Pyle para sexo masculino.
A cromatografia plana e bidimensional de aminoácidos em papel não revelou elevação na excreção urinária de aminoácidos. Níveis séricos de aminoácidos mostraram-se dentro dos padrões de normalidade. Não foi detectado indican urinário, tampouco qualquer outra alteração durante realização de exames de rastreamento para erros inatos do metabolismo. Não há na família relato de quadro clínico semelhante ou distúrbios sugestivos de doença metabólica específica.
Paciente permaneceu internado durante 11 dias em uso de complexo polivitamínico B (80mg/dia de nicotinamida). Com melhora progressiva do quadro cutâneo e neurológico a partir do quarto dia de internação, recebeu alta com marcha atípica e resolução quase completa da dermatite pelagróide. Nos três meses subseqüentes retornou ao ambulatório de pediatria, assintomático e em uso de complexo polivitamínico B (20mg/dia de nicotinamida).
DISCUSSÃO
As manifestações clínicas de dermatite pelagróide – caracterizada por lesões eritêmato-descamativas em áreas de exposição ao sol – associadas ao surgimento concomitante de ataxia cerebelar intermitente, atingindo a inabilidade para deambulação, enquadram perfeitamente o paciente descrito ao diagnóstico clínico de doença de Hartnup em sua apresentação clássica. Além disso, achados secundários, tais como labilidade emocional, hiper-reflexia global de tendões profundos, crise convulsiva e alterações difusas no EEG, foram citados por outros autores como possíveis achados em pacientes com doença de Hartnup.
O caráter intermitente das alterações clínicas com fases de exacerbação verificadas principalmente no período de junho-julho, idêntico ao relatado por Da Gloria e cols., muito provavelmente, segundo relato da mãe, deve-se, nesse caso, à significativa exposição do paciente à luz solar nesse período, coincidente com o de férias escolares, ao que se soma o fato de as estações do ano serem pouco definidas na região.
Goulon e cols.5 em seu trabalho subdividem os casos de pelagra em exógenos e endógenos. A pelagra exógena está relacionada à hipovitaminose B3 induzida principalmente por dieta pobre em triptofano e não está associada a aumento da excreção renal de aminoácidos, ao contrário da pelagra endógena, cujo principal exemplo é a doença de Hartnup, sendo a queda dos níveis de niacina resultado de defeito na absorção renal e intestinal do triptofano.
Pelagra carencial deve ser considerada o principal diagnóstico diferencial, afastado, no entanto, pela ausência dos distúrbios gastrointestinais típicos da síndrome (em especial a diarréia) e pela diferença entre as manifestações neuropsíquicas apresentadas pelo paciente e a demência característica da pelagra exógena. Além disso, a ingesta diária de leite de vaca (alimento rico em triptofano), a sazonalidade das alterações clínicas e a não-observância de sinais e sintomas semelhantes em outras crianças da comunidade rural habitada pelo paciente são também fatores que se somam para afastar o diagnóstico de pelagra nutricional. Outras etiologias possíveis para a pelagra endógena, além da doença de Hartnup, seriam o uso prolongado de isoniazida, de 6-mercaptopurina e tumores carcinóides, situações essas não verificadas no paciente em questão.5
Assim, frente ao quadro clínico apresentado pelo paciente, somado à eficácia do tratamento proposto, responsável pela remissão tanto de lesões cutâneas como das alterações neurológicas, sugere-se diagnóstico de pelagra endógena. Porém, tal como nos casos descritos por Goulon e cols. (França), Da Gloria e cols.(Brasil), Borrie e Lewis e Tada e cols. (Japão), neste também não se evidenciou hiperaminoacidúria típica da doença de Hartnup quando da realização da cromatografia plana e bidimensional de aminoácidos em papel.
Possível explicação já aventada para a não-verificação de aminoacidúria em pacientes com quadro clínico da doença de Hartnup seria uma alteração no metabolismo do triptofano que acarretaria distúrbio em sua transformação em cirunenina por defeito da enzima triptofano-pirrolase. Ou, ainda, como no caso descrito por Borrie e Lewis,a dicotomia entre quadro clínico e achados laboratoriais poderia ser devida a possível genótipo heterozigoto do paciente avaliado.
Entretanto, assim como os casos apresentados por Goulon e cols. e Da Gloria e cols., não foi possível precisar se o déficit de niacina seria resultado de alteração no metabolismo ou transporte renal/intestinal do triptofano. Da Gloria e cols. questionam se a presença de sinais e sintomas da doença de Hartnup sem associação com qualquer disfunção metabólica até agora identificada ou com níveis aumentados de aminoácidos na urina poderia até mesmo tratar-se de uma nova entidade, o que, acreditam os autores, deve ser alvo de investigação, dadas a singularidade e a semelhança do caso aqui descrito com os demais citados."
ANTES ANTES/DEPOIS


Fonte bibliográfica: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0365-05962006000500009&lng=pt&nrm=iso
Baseando-se no artigo, podemos ver que a pelagra pode apresentar um caráter endógeno, conhecido como Doença de Hartnup, sendo caracterizada pela má absorção de triptofano nos rins e intestino delgado, causando uma queda nos níveis de niacina no organismo. Entretanto, comparar pelagra com a Doença de Hartnup não é interessante, pois a pelagra é ocasionada pela deficiência na ingestão de vitamina B3, triptofano e a Doença de Hartnup é consequência de um defeito genético que pode acarretar um quadro clínico semelhante ao de pelagra, devido a baixa concentração de niacina no metabolismo. Além disso, os sintomas são diferenciados, não há uma definição precisa da Doença de Hartnup, não há conclusões nas investigações desta doença.
By: Ivana Melo
Participações do NAD nas Vias Metabólicas
GLICÓLISE: nesta via o NAD funciona como receptor de hirogênio, tornando-se reduzido na forma NADH, conservando em si altas concentrações de energia que serão utilizadas numa outra etapa da via.

CICLO DE KREBS: ocorre na matriz mitocôndrial, o NAD primeiramente funciona como receptor de elétrons e prótons de hidrogênio, sendo reduzido a NADH, que portará a energia liberada pelas oxidações. Em uma outra etapa (CTE) o NADH é oxidado, liberando H+, esse próton vai se unir a uma molécula de O2, formando água.
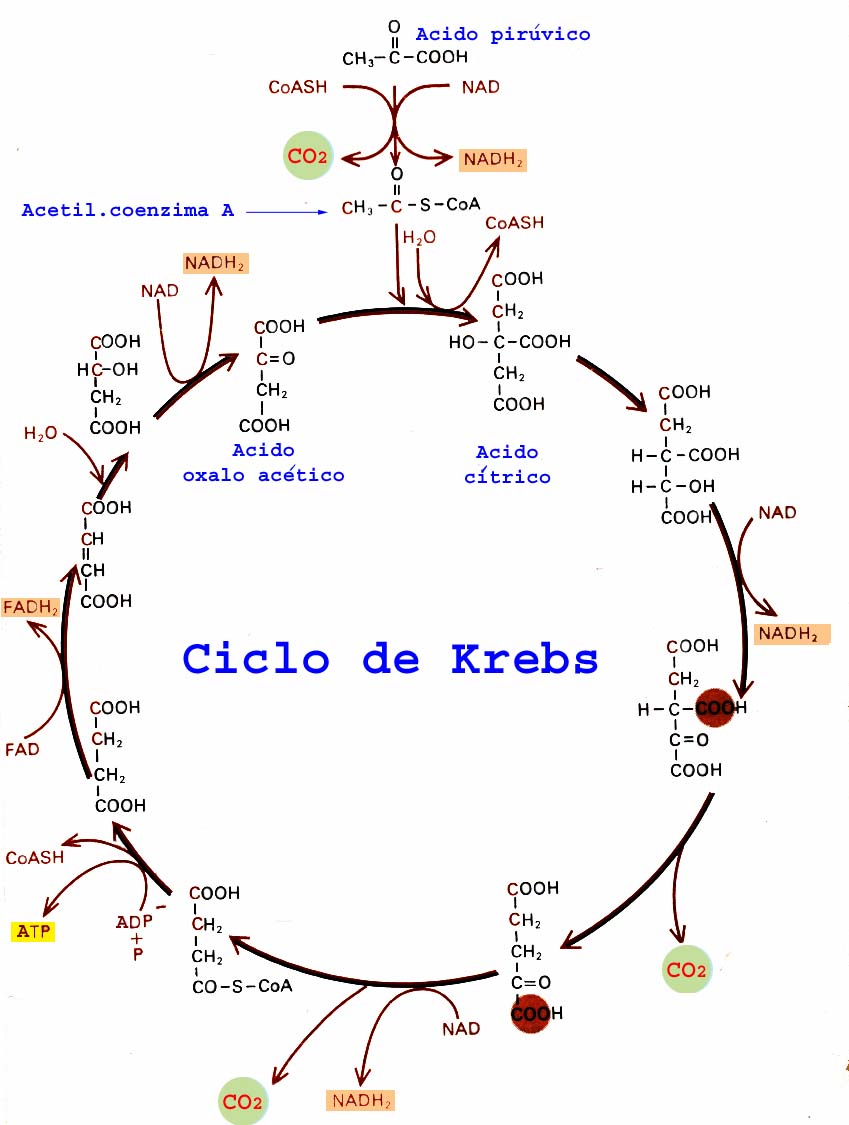
GLICONEOGÊNESE: o malato que está presente dentro da mitocôndria é tranasportado para o citossol, lá ele é reoxidado formando oxaloacetato com produção de NADH. Sem NADH para liberar energia na conversão de 1,3-bifosfoglicerato em gliceraldeído 3-fosfato, não há síntese de glicose, logo a oxidação de NADH a NAD é extremamente importante para tal via. É importante durante uma situação de jejum prolongao.
By: Ivana Melo
CICLO DE KREBS: ocorre na matriz mitocôndrial, o NAD primeiramente funciona como receptor de elétrons e prótons de hidrogênio, sendo reduzido a NADH, que portará a energia liberada pelas oxidações. Em uma outra etapa (CTE) o NADH é oxidado, liberando H+, esse próton vai se unir a uma molécula de O2, formando água.
GLICONEOGÊNESE: o malato que está presente dentro da mitocôndria é tranasportado para o citossol, lá ele é reoxidado formando oxaloacetato com produção de NADH. Sem NADH para liberar energia na conversão de 1,3-bifosfoglicerato em gliceraldeído 3-fosfato, não há síntese de glicose, logo a oxidação de NADH a NAD é extremamente importante para tal via. É importante durante uma situação de jejum prolongao.
By: Ivana Melo
quinta-feira, 20 de novembro de 2008
Niacina
De acordo com o artigo "Farmacologia da niacina ou ácido nicotínico", a niacina uma vitamina solúvel com propriedades hipolipemiantes reduz triglicérides (20% - 50%), LDL (5% - 25%), e aumenta HDL (15% - 35%) O estudo Coronary Drug Projectmostrou que o uso de niacina foi associado com redução de eventos coronários e mortalidade total, e mais recentemente, foi demonstrado que niacina combinada com outras drogas hipolipemiantes pode atenuar a progressão
da aterosclerose coronária. A niacina parece reduzir a mobilização de ácidos graxos livres dos adipócitos, agindo em receptores específicos, diminuindo a formação de lipoproteínas ricas em triglicérides pelo fígado. Porém, a ação da niacina não é totalmente conhecida.
A niacina é metabolizada por duas vias metabólicas: a via na qual é conjugada com a glicina para formaçãodo ácido nicotinúrico e a via que abrange uma série de reações de óxido-redução que formam a nicotinamida e derivados pirimidínicos.
da aterosclerose coronária. A niacina parece reduzir a mobilização de ácidos graxos livres dos adipócitos, agindo em receptores específicos, diminuindo a formação de lipoproteínas ricas em triglicérides pelo fígado. Porém, a ação da niacina não é totalmente conhecida.
A niacina é metabolizada por duas vias metabólicas: a via na qual é conjugada com a glicina para formaçãodo ácido nicotinúrico e a via que abrange uma série de reações de óxido-redução que formam a nicotinamida e derivados pirimidínicos.
Atualmente existem três formulações do ácido nicotínico: liberação imediata ou cristalina, liberação intermediária, prolongada também conhecida com extendida na literatura de língua inglesa e a forma de liberação lenta. A niacina de liberação imediata é rapidamente absorvida
e excretada e, geralmente, é prescrita em múltiplas doses. A niacina de liberação lenta tem um tempo de dissolução geralmente maior que 12 horas. Entre as duas formas clássicas de liberação da niacina encontra-se a forma de liberação intermediária ou extendida. A mesma é absorvida num período de 8 a 12 horas. A mesma deve ser ingerida uma vez por dia e é a única
formulação aprovada pelo FDA para tratamento das dislipidemias. A niacina de liberação imediata rapidamente satura a via da nicotinamida sendo preferencialmente metabolizada pela via do ácido nicotinúrico, fato que leva a alta prevalência de “flushing” (calor e rubor e causado pela liberação de prostaglandinas durante a formação do ácido nicotinúrico) e ausência de hepatotoxicidade. Já a formulação de ação lenta raramente
causa rubor devido a sua preferência pela via da nicotinamida.
Por outro lado, essa formulação associa-se
alto risco de hepatotoxicidade dose dependente. A formulação de liberação intermediária fica num meio termo entre as duas outras formulações sendo que apresenta cerca de 60% menos rubor que a formulação de liberação imediata e raramente apresenta toxicidade hepática em doses de até 2 g/dia.
O uso da niacina de liberação rápida é limitado pela alta taxa de efeitos colaterais (10-50% dos casos de interrupção do tratamento): rubor, calor, prurido, náusea, dispepsia, dor abdominal e diarréia. Em comparação com a formulação de liberação rápida a forma lenta não causa “flushing”, contudo, cerca de 75% dos participantes de estudos randomizados apresentaram aumento das aminotrasnferases três vezes o limite superior do normal, sendo que muitos apresentaram sinais de insuficiência hepática. Por outro lado a niacina de liberação intermediária
causa “flushing” em cerca de 50% dos casos, efeito colateral que vai diminuindo com o passar do tempo. Em estudos controlados apenas 5% dos pacientes suspenderam o tratamento devido a esse fato. O aumento das aminotrasnferases duas vezes acima do limite superior do normal ocorreu em apenas 2,6% dos casos, geralmente em associação com estatinas ou colestiramina e foi reversível com a suspensão dos medicamentos. O uso dessa formulação não se associa a insuficiência hepática.
Um efeito colateral descrito com o uso da niacina de liberação imediata é a hiperglicemia. Esse fato é preocupante, pois uma grande parcela de portadores de diabetes e resistência à insulina tem níveis baixos de HDL-C sendo que a niacina seria uma ótima opção terapêutica. Contudo, há evidência de que com a formulação de liberação intermediária as alterações da glicemia são discretas e transitórias.
Outros possíveis e raros efeitos colaterais com as diversas formulações de niacina são a hiperuricemia, gota, arritmias cardíacas, tontura, calafrios, edema, cefaléia, e ativação de úlcera péptica.
Uma outra possibilidade é a toxicidade muscular (CPK > 10 vezes o limite superior do normal) com o uso isolado, mas principalmente com a associação niacina-estatinas. A incidência desses casos é muito baixa e acredita-se que ocorra na ordem de 2-4/1000. 10 De 601 casos avaliados pelo FDA em 2002 apenas 4 (0,7%) ocorreram pela associação.

O artigo completo está aqui:
http://www.scielo.br/pdf/abc/v85s5/v85s5a05.pdf
e excretada e, geralmente, é prescrita em múltiplas doses. A niacina de liberação lenta tem um tempo de dissolução geralmente maior que 12 horas. Entre as duas formas clássicas de liberação da niacina encontra-se a forma de liberação intermediária ou extendida. A mesma é absorvida num período de 8 a 12 horas. A mesma deve ser ingerida uma vez por dia e é a única
formulação aprovada pelo FDA para tratamento das dislipidemias. A niacina de liberação imediata rapidamente satura a via da nicotinamida sendo preferencialmente metabolizada pela via do ácido nicotinúrico, fato que leva a alta prevalência de “flushing” (calor e rubor e causado pela liberação de prostaglandinas durante a formação do ácido nicotinúrico) e ausência de hepatotoxicidade. Já a formulação de ação lenta raramente
causa rubor devido a sua preferência pela via da nicotinamida.
Por outro lado, essa formulação associa-se
alto risco de hepatotoxicidade dose dependente. A formulação de liberação intermediária fica num meio termo entre as duas outras formulações sendo que apresenta cerca de 60% menos rubor que a formulação de liberação imediata e raramente apresenta toxicidade hepática em doses de até 2 g/dia.
O uso da niacina de liberação rápida é limitado pela alta taxa de efeitos colaterais (10-50% dos casos de interrupção do tratamento): rubor, calor, prurido, náusea, dispepsia, dor abdominal e diarréia. Em comparação com a formulação de liberação rápida a forma lenta não causa “flushing”, contudo, cerca de 75% dos participantes de estudos randomizados apresentaram aumento das aminotrasnferases três vezes o limite superior do normal, sendo que muitos apresentaram sinais de insuficiência hepática. Por outro lado a niacina de liberação intermediária
causa “flushing” em cerca de 50% dos casos, efeito colateral que vai diminuindo com o passar do tempo. Em estudos controlados apenas 5% dos pacientes suspenderam o tratamento devido a esse fato. O aumento das aminotrasnferases duas vezes acima do limite superior do normal ocorreu em apenas 2,6% dos casos, geralmente em associação com estatinas ou colestiramina e foi reversível com a suspensão dos medicamentos. O uso dessa formulação não se associa a insuficiência hepática.
Um efeito colateral descrito com o uso da niacina de liberação imediata é a hiperglicemia. Esse fato é preocupante, pois uma grande parcela de portadores de diabetes e resistência à insulina tem níveis baixos de HDL-C sendo que a niacina seria uma ótima opção terapêutica. Contudo, há evidência de que com a formulação de liberação intermediária as alterações da glicemia são discretas e transitórias.
Outros possíveis e raros efeitos colaterais com as diversas formulações de niacina são a hiperuricemia, gota, arritmias cardíacas, tontura, calafrios, edema, cefaléia, e ativação de úlcera péptica.
Uma outra possibilidade é a toxicidade muscular (CPK > 10 vezes o limite superior do normal) com o uso isolado, mas principalmente com a associação niacina-estatinas. A incidência desses casos é muito baixa e acredita-se que ocorra na ordem de 2-4/1000. 10 De 601 casos avaliados pelo FDA em 2002 apenas 4 (0,7%) ocorreram pela associação.

O artigo completo está aqui:
http://www.scielo.br/pdf/abc/v85s5/v85s5a05.pdf
Por: Pollana Roberta Alves Campos
terça-feira, 18 de novembro de 2008
Epidemia de pelagra na Angola
O artigo a seguir fala sobre uma epidemia de pelagra na Angola, lá as pessoas são extremamente pobres e têm uma alimentação bastante precária, que se tornou ainda mais difícil num momento em que o país passou por uma guerra civil. Mesmo em Inglês, o artigo é muito interessante, vale a pena ler.
Pellagra outbreak in Kuito, Angola
Sophie Baquet, François Wuillaume, Kathia Van Egmond and Felicitas Ibañez
"Since late 1998, over 100000 people living in Bie province, Central Angola, have fled the armed conflict and sought asylum in camps around Kuito, the provincial capital city. Most of the displaced people (estimated 110 000 people) depend entirely on the World Food Programme (WFP) food distribution. The resident population of about 135 000 is also badly effected by the enclavement of one of the most heavily mined cities in the world. Small agricultural fields and seeds have been distributed to the displaced population but the next harvest due about now is expected to be insufficient.
A pellagra outbreak started in Kuito in July, 1999. From July, 1999, until mid-February, 2000, 908 cases of pellagra were recorded at the hospital. The definition used is a dermatitis on two different and symmetrical sites exposed to sunlight or with a typical casal's necklace.
The entire pellagra attack rate is 3·6/1000; 66% of the patients registered with pellagra were displaced people, 83% were women, and 18% of the patients were younger than age 15 years. No data are available to estimate the case fatality. The current attack rate is most probably an underestimate, because no active case searching is done. Also, the case definition used means that only typical and therefore advanced cases are diagnosed. The classical skin lesions seem to occur in older people, and people who do not go outside do not get the skin lesions either. In truth, most of the population probably have a certain degree of niacin deficiency.
From April, 1999, to February, 2000, food rations distributed principally to the displaced population provided an average of 8 mg niacin per person per day. WHO recommended daily intake of niacin equivalents is 6·6 mg (infants) to 21·1 mg (adults).1 WHO technical report Series 362. Requirements of vitamin A, thiamine, riboflavine and niacin. Report of a Joint FAO/WHO expert group. WHO: Geneva, 1967.1 Fish has been distributed since November, 1999, but solely to beneficiaries of supplementary feeding programmes and those diagnosed with pellagra.
In the absence of an appropriate response, Medecins Sans Frontieres (MSF) organised a distribution of vitamin B complex tablets to all women aged 15 years and over in December, 1999. This targeting was done because lack of resources to organise a distribution to the entire population, which was started at the end of February. This outbreak confirms previous observations that people depending on insufficient food rations (in quantity and quality) suffer from different deficiencies.2, 3 and 4 Epicentre and MSF described a similar outbreak of pellagra among Mozambican refugees in Malawi in 1990.5
Despite official international nutrition recommendations, relief programmes fail in the provision of the minimum recommended daily allowance of essential micronutrients. This pellagra outbreak in Kuito underscores the vulnerability of food-aid dependent populations to such forgotten micronutrient deficiency diseases. An urgent effort to supply niacin-rich (ground-nuts, blended food, or fortified food) to this food-aid-dependent population, has to be done rapidly to stem this outbreak, which started 10 months ago."
References
1 WHO technical report Series 362. Requirements of vitamin A, thiamine, riboflavine and niacin. Report of a Joint FAO/WHO expert group. WHO: Geneva, 1967.
2 AM Magan, M Warsame, AK Ali-Salad and M Toole, An outbreak of scurvy in Somali refugees camps, Disasters 7 (1982), pp. 93–97.
3 J Seaman and JPW Rivers, Scurvy and anaemia in refugees, Lancet 1 (1989), p. 1204. Abstract | View Record in Scopus | Cited By in Scopus (3)
4 A Berry-Koch, R Moench, P Hakewell and M Dualeh, Alleviation of nutritional deficiency diseases in refugees. Food Nutr, Bull 12 (1990), pp. 106–112. View Record in Scopus | Cited By in Scopus (5) "
Fonte:
http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T1B-4FVCHYK-5S&_user=687355&_coverDate=05%2F20%2F2000&_alid=823308986&_rdoc=19&_fmt=high&_orig=search&_cdi=4886&_sort=d&_docanchor=&view=c&_ct=236&_acct=C000037918&_version=1&_urlVersion=0&_userid=687355&md5=9b91ef6b37d71b1ccb87516ba7ecf98c
Pellagra outbreak in Kuito, Angola
Sophie Baquet, François Wuillaume, Kathia Van Egmond and Felicitas Ibañez
"Since late 1998, over 100000 people living in Bie province, Central Angola, have fled the armed conflict and sought asylum in camps around Kuito, the provincial capital city. Most of the displaced people (estimated 110 000 people) depend entirely on the World Food Programme (WFP) food distribution. The resident population of about 135 000 is also badly effected by the enclavement of one of the most heavily mined cities in the world. Small agricultural fields and seeds have been distributed to the displaced population but the next harvest due about now is expected to be insufficient.
A pellagra outbreak started in Kuito in July, 1999. From July, 1999, until mid-February, 2000, 908 cases of pellagra were recorded at the hospital. The definition used is a dermatitis on two different and symmetrical sites exposed to sunlight or with a typical casal's necklace.
The entire pellagra attack rate is 3·6/1000; 66% of the patients registered with pellagra were displaced people, 83% were women, and 18% of the patients were younger than age 15 years. No data are available to estimate the case fatality. The current attack rate is most probably an underestimate, because no active case searching is done. Also, the case definition used means that only typical and therefore advanced cases are diagnosed. The classical skin lesions seem to occur in older people, and people who do not go outside do not get the skin lesions either. In truth, most of the population probably have a certain degree of niacin deficiency.
From April, 1999, to February, 2000, food rations distributed principally to the displaced population provided an average of 8 mg niacin per person per day. WHO recommended daily intake of niacin equivalents is 6·6 mg (infants) to 21·1 mg (adults).1 WHO technical report Series 362. Requirements of vitamin A, thiamine, riboflavine and niacin. Report of a Joint FAO/WHO expert group. WHO: Geneva, 1967.1 Fish has been distributed since November, 1999, but solely to beneficiaries of supplementary feeding programmes and those diagnosed with pellagra.
In the absence of an appropriate response, Medecins Sans Frontieres (MSF) organised a distribution of vitamin B complex tablets to all women aged 15 years and over in December, 1999. This targeting was done because lack of resources to organise a distribution to the entire population, which was started at the end of February. This outbreak confirms previous observations that people depending on insufficient food rations (in quantity and quality) suffer from different deficiencies.2, 3 and 4 Epicentre and MSF described a similar outbreak of pellagra among Mozambican refugees in Malawi in 1990.5
Despite official international nutrition recommendations, relief programmes fail in the provision of the minimum recommended daily allowance of essential micronutrients. This pellagra outbreak in Kuito underscores the vulnerability of food-aid dependent populations to such forgotten micronutrient deficiency diseases. An urgent effort to supply niacin-rich (ground-nuts, blended food, or fortified food) to this food-aid-dependent population, has to be done rapidly to stem this outbreak, which started 10 months ago."
References
1 WHO technical report Series 362. Requirements of vitamin A, thiamine, riboflavine and niacin. Report of a Joint FAO/WHO expert group. WHO: Geneva, 1967.
2 AM Magan, M Warsame, AK Ali-Salad and M Toole, An outbreak of scurvy in Somali refugees camps, Disasters 7 (1982), pp. 93–97.
3 J Seaman and JPW Rivers, Scurvy and anaemia in refugees, Lancet 1 (1989), p. 1204. Abstract | View Record in Scopus | Cited By in Scopus (3)
4 A Berry-Koch, R Moench, P Hakewell and M Dualeh, Alleviation of nutritional deficiency diseases in refugees. Food Nutr, Bull 12 (1990), pp. 106–112. View Record in Scopus | Cited By in Scopus (5) "
Fonte:
http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T1B-4FVCHYK-5S&_user=687355&_coverDate=05%2F20%2F2000&_alid=823308986&_rdoc=19&_fmt=high&_orig=search&_cdi=4886&_sort=d&_docanchor=&view=c&_ct=236&_acct=C000037918&_version=1&_urlVersion=0&_userid=687355&md5=9b91ef6b37d71b1ccb87516ba7ecf98c
Por: Pollana Roberta Alves Campos
domingo, 16 de novembro de 2008
Relação entre a pelagra e a anorexia
De acordo com o artigo: "Pellagra May Be a Rare SecondaryComplication of Anorexia Nervosa: A Systematic Review of the Literature", a literatura revela que vários casos de pessoas com anorexia que apresentavam pelagra. As características mais comuns da pelagra em pacientes com anorexia são manifestações cutâneas tais como eritema em áreas expostas ao sol; glossite e estomatite.
Uma análise feita por Patrick, discutiu sobre a etiologia das doenças nutricionais associadas à anorexia, bulimia e transtornos alimentares atípicos. O artigo completo, porém, não relacionou a eventual ligação entre a pelagra e a anorexia. Nesse artigo, quatro casos de pelagra em pacientes com anorexia nervosa da literatura são revisados.
Bicknell e Prescott descreveram a história detalhada da pelagra. A palavra pelagra significa pele áspera e foi escrita por Frapolli em 1771. Ele foi o primeiro a notar lesões cutâneas em resposta à exposição à luz solar. A doença foi identificada primeiramente na Itália e Espanha, mas logo foi encontrada em outros países da Europa. Apesar de ser descrita inicialmente na América do Norte em 1864, a pelagra se fez presente antes dessa data. Em 1937, a língua preta foi descoberta (esta é uma doença semelhante à pelagra, porém ocorre em caninos) e foi curada com o uso de niacina. Algum tempo depois, a niacina foi usada em humanos com bons resultados, mas não completamente curativos. Em 1947 a complexidade da pelagra foi resolvida. Verificou-se que o aminoácido essencial triptofano, um precursor metabólico da niacina , poderia curar ou prevenir a pelagra. Em 1950, estudos demonstraram que 60 mg de triptofano resultavam em 1 mg de niacina. Considerando que a dieta ocidental típica fornece aproximadamente 1 g de triptofano diariamente a partir de fontes de proteína e de outros alimentos, as exigências diárias de niacinas podem ser satisfeitas a partir da dieta somente. Assim determinou-se que a melhor forma de tratar ou prevenir a pelagra era administrar niacina ou assegurar uma ingestão adequada de proteínas. Embora a pelagra seja caracterizada pelos 4 D's, os sintomas iniciais odem ser: anorexia, ansiedade, psicose, delírio, dermatite, baixa resistência, melancolia, náusea, perda de peso e vômito.
De acordo com o artigo, alterações cutâneas estiveram entre os principais sintomas da pelagra em pacientes com anorexia nervosa. Considerando a complexidade a anorexia nervosa e as muitas manifestações clínicas da doença, é difícil determinar quais sintomas são reflexivos de pelagra. Deve-se suspeitar que o paciente apresenta pelagra quando a história do paciente anoréxico revela manifestações cutâneas relacionadas com exposição à luz solar. Para confirmar uma suspeita clínica de pelagra, um profissional de saúde pode tratar o paciente com 150-500 mg de niacina dividida em doses diárias, e acompanhar melhorias cutâneas 24-48 horas depois. Se houver melhorias cutâneas, o diagnóstico de pelagra pode ser feito.
Quando há uma ingestão de niacina e/ou de protéinas inadequada, ocorre uma perda da inibição do feedback sobre o percurso kynurenine , desviando mais triptofano para o percurso kynurenine , fazendo com que hajam menos substrato disponível para a síntese de serotonina e
resultando em uma diminuição dos níveis de 5-HIAA (5-hydroxy-indole-acetic acid) urinário.
Além de uma deficiente ingestão de niacina e/ou proteína, outros fatores podem levar à características cutâneas e/ou quadro clínico de pelagra em pacientes anoréxicos. Por exemplo, a deficiência de riboflavina e/ou piridoxina pode comprometer a biossíntese de niacina a partir de triptofano. A riboflavina é a coenzima para kynurenine hidroxilase e a piridoxina é a coenzima para a kynureninase - ambas as enzimas permitem a conversão de triptofano in vivo. Se há deficiência de niacina ou de qualquer uma dessas vitaminas, existe a possibilidade de desenvolvimento de quadro clínico de pelagra.
Fatores tais como excessiva ingestão de leucina (o que não seria visto em pacientes anoréxicos), estrogênios e progestogênios, síndrome carcinóide e diversas medicações poderiam também levar ao desenvolvimento de pelagra. Estrogênios são inibidores competitivos da kynureninase, enquanto progestogênios são podem reduzir a atividade da kynurenine hydroxylase. A síndrome carcinóide, um tumor nas células do trato intestinal, desvia a maior parte de triptofano da biossíntese de NAD e NADP para a síntese de serotonina. Finalmente, medicações como Isoniazida, Carbidopa, estão associadas com o esgotamento de niacina.

Bibliografia:
PROUSKY, E. Jonathan. ND, FRSH. Pellagra May Be a Rare SecondaryComplication of Anorexia Nervosa:A Systematic Review of the Literature. Alternative Medicine Review
Uma análise feita por Patrick, discutiu sobre a etiologia das doenças nutricionais associadas à anorexia, bulimia e transtornos alimentares atípicos. O artigo completo, porém, não relacionou a eventual ligação entre a pelagra e a anorexia. Nesse artigo, quatro casos de pelagra em pacientes com anorexia nervosa da literatura são revisados.
Bicknell e Prescott descreveram a história detalhada da pelagra. A palavra pelagra significa pele áspera e foi escrita por Frapolli em 1771. Ele foi o primeiro a notar lesões cutâneas em resposta à exposição à luz solar. A doença foi identificada primeiramente na Itália e Espanha, mas logo foi encontrada em outros países da Europa. Apesar de ser descrita inicialmente na América do Norte em 1864, a pelagra se fez presente antes dessa data. Em 1937, a língua preta foi descoberta (esta é uma doença semelhante à pelagra, porém ocorre em caninos) e foi curada com o uso de niacina. Algum tempo depois, a niacina foi usada em humanos com bons resultados, mas não completamente curativos. Em 1947 a complexidade da pelagra foi resolvida. Verificou-se que o aminoácido essencial triptofano, um precursor metabólico da niacina , poderia curar ou prevenir a pelagra. Em 1950, estudos demonstraram que 60 mg de triptofano resultavam em 1 mg de niacina. Considerando que a dieta ocidental típica fornece aproximadamente 1 g de triptofano diariamente a partir de fontes de proteína e de outros alimentos, as exigências diárias de niacinas podem ser satisfeitas a partir da dieta somente. Assim determinou-se que a melhor forma de tratar ou prevenir a pelagra era administrar niacina ou assegurar uma ingestão adequada de proteínas. Embora a pelagra seja caracterizada pelos 4 D's, os sintomas iniciais odem ser: anorexia, ansiedade, psicose, delírio, dermatite, baixa resistência, melancolia, náusea, perda de peso e vômito.
De acordo com o artigo, alterações cutâneas estiveram entre os principais sintomas da pelagra em pacientes com anorexia nervosa. Considerando a complexidade a anorexia nervosa e as muitas manifestações clínicas da doença, é difícil determinar quais sintomas são reflexivos de pelagra. Deve-se suspeitar que o paciente apresenta pelagra quando a história do paciente anoréxico revela manifestações cutâneas relacionadas com exposição à luz solar. Para confirmar uma suspeita clínica de pelagra, um profissional de saúde pode tratar o paciente com 150-500 mg de niacina dividida em doses diárias, e acompanhar melhorias cutâneas 24-48 horas depois. Se houver melhorias cutâneas, o diagnóstico de pelagra pode ser feito.
Quando há uma ingestão de niacina e/ou de protéinas inadequada, ocorre uma perda da inibição do feedback sobre o percurso kynurenine , desviando mais triptofano para o percurso kynurenine , fazendo com que hajam menos substrato disponível para a síntese de serotonina e
resultando em uma diminuição dos níveis de 5-HIAA (5-hydroxy-indole-acetic acid) urinário.
Além de uma deficiente ingestão de niacina e/ou proteína, outros fatores podem levar à características cutâneas e/ou quadro clínico de pelagra em pacientes anoréxicos. Por exemplo, a deficiência de riboflavina e/ou piridoxina pode comprometer a biossíntese de niacina a partir de triptofano. A riboflavina é a coenzima para kynurenine hidroxilase e a piridoxina é a coenzima para a kynureninase - ambas as enzimas permitem a conversão de triptofano in vivo. Se há deficiência de niacina ou de qualquer uma dessas vitaminas, existe a possibilidade de desenvolvimento de quadro clínico de pelagra.
Fatores tais como excessiva ingestão de leucina (o que não seria visto em pacientes anoréxicos), estrogênios e progestogênios, síndrome carcinóide e diversas medicações poderiam também levar ao desenvolvimento de pelagra. Estrogênios são inibidores competitivos da kynureninase, enquanto progestogênios são podem reduzir a atividade da kynurenine hydroxylase. A síndrome carcinóide, um tumor nas células do trato intestinal, desvia a maior parte de triptofano da biossíntese de NAD e NADP para a síntese de serotonina. Finalmente, medicações como Isoniazida, Carbidopa, estão associadas com o esgotamento de niacina.
Bibliografia:
PROUSKY, E. Jonathan. ND, FRSH. Pellagra May Be a Rare SecondaryComplication of Anorexia Nervosa:A Systematic Review of the Literature. Alternative Medicine Review
Por: Pollana Roberta Alves Campos

" MANIFESTACIONES CLÍNICAS
Las personas que sufren de pelagra en general parecen pobremente nutridas. A menudo se sienten débiles y tienen poco peso. La enfermedad está caracterizada por «las Tres D»: dermatitis, diarrea y demencia . Se presentan ligeros cambios sensoriales y motores, así como una disminución de la sensibilidad al tacto suave, algo de debilidad muscular y temblor. También se han descrito otros síntomas, pero sin embargo la parálisis es rara. Los casos de pelagra no tratados pueden causar la muerte.
Dermatitis
Frecuentemente la enfermedad se diagnostica por la apariencia de la piel que presenta lesiones características (Foto 31). Las lesiones aparecen en áreas de la piel expuesta a la luz del sol, como la cara, el reverso de las manos, el cuello, los antebrazos y porciones expuestas de las piernas. Esta dermatitis pelagrosa comienza con un aumento de la pigmentación. Las áreas hiperpigmentadas pierden el brillo aceitoso de la piel sana y se vuelven secas, escamosas y eventualmente agrietadas. Casi siempre hay una línea definida de demarcación entre estas lesiones y la piel sana, ya que la parte afectada es áspera al tacto por lo tanto es fácil de identificar. El estado de la piel puede permanecer estático, cicatrizar o empeorar. Si progresa la lesión, a menudo hay una descamación; puede haber grietas y fisuras y a veces, la piel se puede ampollar. Las vesículas contienen un exudado incoloro. Las áreas que han perdido una capa de piel, a veces son brillantes, delgadas y más bien despigmentadas. Todas estas lesiones cutáneas son en general más o menos simétricas.
En sujetos de raza blanca las lesiones de la piel al principio parecen como el eritema de las quemaduras de sol. En individuos blancos y negros, las lesiones de pelagra producen sensación de quemadura y dolor cuando se exponen a los rayos directos del sol, lo mismo que le sucede a una persona de piel muy blanca quemada por el sol. Las lesiones pueden también corresponder con un orificio o agujeros en la ropa que se usa con frecuencia, lo cual puede permitir que la luz solar llegue a la piel. Por ejemplo, el clásico collar de Casal alrededor del cuello y en la parte superior del tórax es el resultado de la acción del sol sobre esta parte del cuerpo en una persona con una camisa de cuello abierto.La lengua y otras partes de la boca a menudo se inflaman o están rojas, delicadas y con apariencia áspera. La estomatitis angular y la queilosis casi siempre asociadas con la carencia de riboflavina se observan frecuentemente.
Diarrea
Los ataques de dolor abdominal, diarrea y otras molestias digestivas, son frecuentes en los casos de pelagra. Se considera que cambios similares a los que se presentan en y alrededor de la boca, están presentes en otras partes del tracto digestivo, y pueden ser la causa del malestar abdominal y de la quemazón intestinal. Pocos de estos síntomas y signos son específicos de la pelagra, pero si se acompañan de cambios en la piel o de síntomas mentales o responden a la niacina, confirman el diagnóstico de pelagra.
Demencia
El compromiso del sistema nervioso se manifiesta por síntomas y signos sumamente variables. Los más comunes son irritabilidad, pérdida de memoria, ansiedad e insomnio. Estos síntomas pueden llevar a la demencia, y en la práctica no es raro que personas con demencia resultante de la pelagra sean admitidas en instituciones mentales. Se deben por lo tanto examinar todos los casos de demencia, sobre todo donde el maíz es el alimento básico y se presenta la pelagra, para buscar otros signos de esta enfermedad."
A figura mostra a principal consequência na falta de niacina (vitamina B3), o famoso " 3D : dermatite, diarréia e demência". Esse sintomas vão aparecendo conforme o grau de carência no organismo. A dermatite e a diarréia são reversíveis a partir do uso de dietas ricas em vitaminas, porém a demência em estado avançado é irreversível, levando a morte.
By: Ivana Melo
Pelagra: diarréia, dermatite e demência
A incidência atual de pelagra nos Estados Unidos e em outros países é desconhecida, epidemias de pelagra não mais evidentes. Casos esporádicos ocorrem entre alcoolátras anôminos, pessoas dependentes de drogas e pacientes com má absorção. Geralmente, é uma doença que ocorre em adultos sem predileção sexual ou racial. Raramente ocorre durante a infância. Adolescentes podem desenvolver pelagra desde que possuam carência nutricional.
A pelagra primária resulta em uma deficiência celular de niacina, resultado de uma baixa ingestão de triptofano e/ou niacina. A niacina é convertida em uma amida no corpo. A nicotinamida é um derivado natural de niacina. A niacina é essencial para o metabolismo celular como um compoente do NAD e NADP, que doam ou recebem íons de hidragênio em reações metabólicas. Esses compostos são coenzimas importantes para a glicólise, metabolismo de proteínas e aminoácidos , metabolismo do piruvato, metabolismo de glicerol e dezenas de outras vias metabólicas. Como muitos orgãos necessitam dessas vias, eles são afetados pela dificiência de niacina. A niacina pode ser obtida diretamente da dieta ou pelo metabolismo do triptofano. 60 mg de triptofano produz 1 mg de niacina na presença de vitaminas B2 e B6. Desse modo, a deficiência de um desses poderia resultar em pelagra em uma pessoa que é nutricionalmente fraca. Da mesma forma, um excesso de leucina na dieta pode interferir com esta
conversão e resultar em pelagra. O recomendado é de 5 a 20 mg diários de niacina equivalentes (dependendo da idade e sexo) com as necessidades adicionais para grávidas e mulheres lactantes.
A pelagra secundária ocorre quando há ingestão adequada de niacina, mas outros fatores ou doenças interferem na digestão e/ou trasformação de niacina, tais como: diarréia prolongada, anorexia nervosa, alcoolismo crônico, tumor carcinóide, síndrome de Hartnup, tuberculose do trato intestinal, entre outros. Em pacientes com tumor carcinóide, o triptofano é convertido em serotonina, diminuindo a produção endógena de niacina. Como a medicação antituberculose é andrógena da niacina, ela pode suprimir a produção endógena de niacina por meio de triptofano. Na doença de Hartnup, os aminoácidos, inclusive o triptofano, são pobremente absorvidos. A administração de 5-fluorouacil é conhecida por causar sintomas da pelagra, já que inibe a conversão de triptofano à niacina. Outras drogas também podem causar pelagra, pois interferem no percurso triptofano-niacina.
A morbidade da pelagra está relacionada aos diversos sistemas inorgânicos envolvidos. Além de dermatite frequente em áreas expostas ao sol, os sintomas são também ulceração da boca e da língua como resultado da membrana mucosa, baixo apetite, náuseas, vômitos, dor abdominal e aumento da salivação. O comprometimento gastrointestinal pode causar mal absorção . Diarréia (resulta do comprometimento da mucosa do trato gastrointestinal), gastrite, úlcera duodenal ocorrem em 50% dos casos. Anorexia, mal absorção e diarréia podem levar a um estado de desnutrição. Alterações neuropsiquiátricas incluem cefaléia, irritabilidade, falta de concentração, anciedade, delírios, alucinações, apatia, fatia, depressão. Fadiga e insônia levam a encefalopatia caracterizada por confusão, perda de memória, e psicose. As alterações no sistema nervoso podem ser encontradas no cérebro, na medula espinhal e em nervos periféricos, essas alterações são caracterizadas pela desmielinização e degeneração do sistema nervoso.
Não existem exames químicos para diagnosticar definitivamente a pelagra, porém o diagnóstico é feito pela análise dos níveis séricos de niacina, triptofano, NAD e NADP.

Fonte:
http://www3.interscience.wiley.com/cgi-bin/fulltext/118749389/HTMLSTART
A pelagra primária resulta em uma deficiência celular de niacina, resultado de uma baixa ingestão de triptofano e/ou niacina. A niacina é convertida em uma amida no corpo. A nicotinamida é um derivado natural de niacina. A niacina é essencial para o metabolismo celular como um compoente do NAD e NADP, que doam ou recebem íons de hidragênio em reações metabólicas. Esses compostos são coenzimas importantes para a glicólise, metabolismo de proteínas e aminoácidos , metabolismo do piruvato, metabolismo de glicerol e dezenas de outras vias metabólicas. Como muitos orgãos necessitam dessas vias, eles são afetados pela dificiência de niacina. A niacina pode ser obtida diretamente da dieta ou pelo metabolismo do triptofano. 60 mg de triptofano produz 1 mg de niacina na presença de vitaminas B2 e B6. Desse modo, a deficiência de um desses poderia resultar em pelagra em uma pessoa que é nutricionalmente fraca. Da mesma forma, um excesso de leucina na dieta pode interferir com esta
conversão e resultar em pelagra. O recomendado é de 5 a 20 mg diários de niacina equivalentes (dependendo da idade e sexo) com as necessidades adicionais para grávidas e mulheres lactantes.
A pelagra secundária ocorre quando há ingestão adequada de niacina, mas outros fatores ou doenças interferem na digestão e/ou trasformação de niacina, tais como: diarréia prolongada, anorexia nervosa, alcoolismo crônico, tumor carcinóide, síndrome de Hartnup, tuberculose do trato intestinal, entre outros. Em pacientes com tumor carcinóide, o triptofano é convertido em serotonina, diminuindo a produção endógena de niacina. Como a medicação antituberculose é andrógena da niacina, ela pode suprimir a produção endógena de niacina por meio de triptofano. Na doença de Hartnup, os aminoácidos, inclusive o triptofano, são pobremente absorvidos. A administração de 5-fluorouacil é conhecida por causar sintomas da pelagra, já que inibe a conversão de triptofano à niacina. Outras drogas também podem causar pelagra, pois interferem no percurso triptofano-niacina.
A morbidade da pelagra está relacionada aos diversos sistemas inorgânicos envolvidos. Além de dermatite frequente em áreas expostas ao sol, os sintomas são também ulceração da boca e da língua como resultado da membrana mucosa, baixo apetite, náuseas, vômitos, dor abdominal e aumento da salivação. O comprometimento gastrointestinal pode causar mal absorção . Diarréia (resulta do comprometimento da mucosa do trato gastrointestinal), gastrite, úlcera duodenal ocorrem em 50% dos casos. Anorexia, mal absorção e diarréia podem levar a um estado de desnutrição. Alterações neuropsiquiátricas incluem cefaléia, irritabilidade, falta de concentração, anciedade, delírios, alucinações, apatia, fatia, depressão. Fadiga e insônia levam a encefalopatia caracterizada por confusão, perda de memória, e psicose. As alterações no sistema nervoso podem ser encontradas no cérebro, na medula espinhal e em nervos periféricos, essas alterações são caracterizadas pela desmielinização e degeneração do sistema nervoso.
Não existem exames químicos para diagnosticar definitivamente a pelagra, porém o diagnóstico é feito pela análise dos níveis séricos de niacina, triptofano, NAD e NADP.

Fonte:
http://www3.interscience.wiley.com/cgi-bin/fulltext/118749389/HTMLSTART
Por: Pollana Roberta Alves Campos

Um pouquinho de história
Historicamente, após a importação de milho para a Europa a partir do Novo Mundo no século 18, verificou-se que surgiu na Espanha como uma epidemia com o nome popular de "Mal de la Rosa" quando Gaspar Casal descreveu esta doença pela primeira vez em 1735. Casal notou uma clara a relação entre o uso do milho, a pobreza e a falta de carne. O primeiro caso descrito nos Estados Unidos foi em 1902, mas em poucos anos a pelagra tornou-se uma epidemia. O algodão era barato, os eram salários baixos e o preço dos alimentos era bastante elevado. O milho era a base da alimentação e, em particular nas áreas urbanas, era moído, e assim, os nutrientes eram removidos. O milho, naturalmente, possui baixo valor nutritivo de nicotinamida e triptofano, e a pouca nicotinamida que possui está em uma forma particularmente indisponível, a menos que o milho seja cozido com produtos alcalinos (é dessa forma que os mexicanos fazem tortilhas, por isso há poucos casos de pelagra no México). O milho moído parecia melhor, mais bem conservado, tornar o pão mais saboroso e era mais conveniente para comprar na loja, mas era nutricionalmente pobre. Em 1920, casos nos estados do sul chegaram a cem mil com até 10000 mortes por ano.
Goldberge reconheceu que a pelagra se tratava de uma doença nutritiva e não um primariamente uma doença infecciosa e que, embora vários indivíduos de uma mesma família apresentassem pelagra, em mais de um terço dos casos ela não foi causada por fatores hereditários. Apesar (ou por causa) da falta de métodos moleculares modernos Goldberger procedeu diretamente para a causa e uma cura completa a preços acessíveis para a pelagra. A adição de leite, ovos e carne para uma dieta institucional e, mais tarde, levedura, impediria que o estado piorasse e até mesmo ajudaria a estabilizar a incidência dos casos. O pressuposto era que uma vitamina era deficiente e foi chamado de G ou vitamina Vitamina PP (preventiva da pelagra). Foi só depois da morte prematura de Goldberger que ela foi identificada como ácido nicotínico e imediatamente aplicada aos humanos. A levedura de cerveja era abundante e fornecida gratuitamente pela Cruz Vermelha Americana. Pobre hábitos alimentares, stress econômico e um grande número de pessoas com desnutrição crônica foi provavelmente a causa de mais 2000 mortes relatadas em 1940. Anos mais tarde a pelagra como uma epidemia desapareceu e se tornou uma rara curiosidade em alcoolistas na América do Norte e esporadicamente em outros locais sob circunstâncias extremas.
A primeira doença neurodegenerativa a ser curada por Goldberger, que foi nomeado para o Prémio Nobel cinco vezes. Focos isolados em todo o mundo ainda ocorrem geralmente na África, Índia e China, normalmente em refusiados de guerra.
A dose diária recomendada (RDA) de nicotinamida é de cerca de 15 mg. A mediana de ingestão, no Reino Unido é de cerca de 35 mg com 1.2% da população tendo até 70 mg / dia. Muito poucos por cento da população ainda estão abaixo da RDA. Um terço da nicotinamida na dieta é proveniente de produtos de carne bovina e, sobretudo, um terço de cereais, do pão e de outras fontes, incluindo café torrado e de hortaliças frescas. Uma pequena percentagem provém das pílulas de vitaminas.
Desde o reconhecimento de ácido nicotínico como o agente preventivo de pelagra, houve um intenso interesse na sua bioquímica. Ele é integrante das co-enzimas NAD e NADP e, portanto, participam de mais de 200 reações enzimáticas. Destaque são as em que há formação de ATP e de diversos neuroprotetores e funções anti-oxidantes. a produção endógena de triptofano é reforçada por piridoxina, riboflavina e tiamina. Seu próprio catabolismo é catalisado pela enzima nicotinamida N-metil transferase (NNMT). Esta enzima é praticamente inexistente nos herbívoros sugerindo que ela foi desenvolvida pela evolução para lidar com a maior ingestão de nicotinamida por onívoros e carnívoros.
Fonte: http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TB9-4H5N272-1&_user=687355&_coverDate=11%2F30%2F2005&_alid=823308986&_rdoc=7&_fmt=high&_orig=search&_cdi=5137&_sort=d&_docanchor=&view=c&_ct=236&_acct=C000037918&_version=1&_urlVersion=0&_userid=687355&md5=7a9dfdf7aea15bc52525dc35310cffb2
Por: Pollana Robeta Alves Campos
Goldberge reconheceu que a pelagra se tratava de uma doença nutritiva e não um primariamente uma doença infecciosa e que, embora vários indivíduos de uma mesma família apresentassem pelagra, em mais de um terço dos casos ela não foi causada por fatores hereditários. Apesar (ou por causa) da falta de métodos moleculares modernos Goldberger procedeu diretamente para a causa e uma cura completa a preços acessíveis para a pelagra. A adição de leite, ovos e carne para uma dieta institucional e, mais tarde, levedura, impediria que o estado piorasse e até mesmo ajudaria a estabilizar a incidência dos casos. O pressuposto era que uma vitamina era deficiente e foi chamado de G ou vitamina Vitamina PP (preventiva da pelagra). Foi só depois da morte prematura de Goldberger que ela foi identificada como ácido nicotínico e imediatamente aplicada aos humanos. A levedura de cerveja era abundante e fornecida gratuitamente pela Cruz Vermelha Americana. Pobre hábitos alimentares, stress econômico e um grande número de pessoas com desnutrição crônica foi provavelmente a causa de mais 2000 mortes relatadas em 1940. Anos mais tarde a pelagra como uma epidemia desapareceu e se tornou uma rara curiosidade em alcoolistas na América do Norte e esporadicamente em outros locais sob circunstâncias extremas.
A primeira doença neurodegenerativa a ser curada por Goldberger, que foi nomeado para o Prémio Nobel cinco vezes. Focos isolados em todo o mundo ainda ocorrem geralmente na África, Índia e China, normalmente em refusiados de guerra.
A dose diária recomendada (RDA) de nicotinamida é de cerca de 15 mg. A mediana de ingestão, no Reino Unido é de cerca de 35 mg com 1.2% da população tendo até 70 mg / dia. Muito poucos por cento da população ainda estão abaixo da RDA. Um terço da nicotinamida na dieta é proveniente de produtos de carne bovina e, sobretudo, um terço de cereais, do pão e de outras fontes, incluindo café torrado e de hortaliças frescas. Uma pequena percentagem provém das pílulas de vitaminas.
Desde o reconhecimento de ácido nicotínico como o agente preventivo de pelagra, houve um intenso interesse na sua bioquímica. Ele é integrante das co-enzimas NAD e NADP e, portanto, participam de mais de 200 reações enzimáticas. Destaque são as em que há formação de ATP e de diversos neuroprotetores e funções anti-oxidantes. a produção endógena de triptofano é reforçada por piridoxina, riboflavina e tiamina. Seu próprio catabolismo é catalisado pela enzima nicotinamida N-metil transferase (NNMT). Esta enzima é praticamente inexistente nos herbívoros sugerindo que ela foi desenvolvida pela evolução para lidar com a maior ingestão de nicotinamida por onívoros e carnívoros.
Fonte: http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TB9-4H5N272-1&_user=687355&_coverDate=11%2F30%2F2005&_alid=823308986&_rdoc=7&_fmt=high&_orig=search&_cdi=5137&_sort=d&_docanchor=&view=c&_ct=236&_acct=C000037918&_version=1&_urlVersion=0&_userid=687355&md5=7a9dfdf7aea15bc52525dc35310cffb2
Por: Pollana Robeta Alves Campos
sexta-feira, 14 de novembro de 2008
Biossíntese de NAD e umas coisinhas a mais
Esse artigo é bem denso, está em inglês, mas é super interessante, trata sobre vários aspectos do NAD, da niacina, inclusive a biossíntese de NAD. É só como curiosidade...
Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition
Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition
Departments of Genetics and Biochemistry and the Norris Cotton Cancer Center, Dartmouth Medical School, Lebanon, New Hampshire 03756;
Abstract
Although baseline requirements for nicotinamide adenine dinucleotide (NAD+) synthesis can be met either with dietary tryptophan or with less than 20 mg of daily niacin, which consists of nicotinic acid and/or nicotinamide, there is growing evidence that substantially greater rates of NAD+ synthesis may be beneficial to protect against neurological degeneration, Candida glabrata infection, and possibly to enhance reverse cholesterol transport. The distinct and tissue-specific biosynthetic and/or ligand activities of tryptophan, nicotinic acid, nicotinamide, and the newly identified NAD+ precursor, nicotinamide riboside, reviewed herein, are responsible for vitamin-specific effects and side effects. Because current data suggest that nicotinamide riboside may be the only vitamin precursor that supports neuronal NAD+ synthesis, we present prospects for human nicotinamide riboside supplementation and propose areas for future research.
| INTRODUCTION |
|---|
Deficiency of niacin and/or tryptophan (Trp) causes pellagra, which is characterized by a darkly pigmented skin rash and the three D's of dermatitis, diarrhea, and dementia. A century ago, pellagra was common among the rural poor in the southern United States and was thought to be an infectious disease. However, in 1914, Joseph Goldberger tested the hypothesis that pellagra might be caused by a dietary deficiency and discovered that substituting corn-based diets with fresh milk, eggs, and meat cured and prevented the condition. Twenty-three years later, Conrad Elvehjem obtained a nicotinamide (Nam) fraction from deproteinized liver and a sample of crystalline nicotinic acid (Na) and showed that these compounds have an antipellagragenic “antiblack tongue” activity on malnourished dogs. Subsequent biochemical studies identified Nam as a component of NAD+ and nicotinamide adenine dinucleotide phosphate (NADP) and showed that animals with pellagra have a significant decrease in muscle and liver NAD+ and NADP. Today pellagra occurs rarely in cases of extreme alcoholism and anorexia, or as a seasonal malady in underdeveloped parts of the world.
As schematized in Figure 1, the reason that a poor diet can produce a requirement for Na and Nam is that Trp, Na, and Nam are all NAD+ precursors. Trp is converted to NAD+ through an eight-step de novo pathway (Figure 2), so termed because the Nam base is essentially made from scratch. In contrast, Na and Nam are considered “salvageable precursors” that require only three steps and two steps, respectively, to rebuild NAD+. Nicotinamide riboside (NR) is an additional salvageable NAD+ precursor vitamin with a two-step pathway and a three-step pathway to form NAD+. As schematized in Figures 1 and 2, cells require ongoing NAD+ synthesis because NAD+ and NADP are not only coenzymes, which are recycled back and forth between oxidized (NAD+ and NADP) and reduced (NADH, NADPH) forms by hydride transfer enzymes, but are also substrates of NAD+-consuming enzymes that break the glycosidic bond between the Nam moiety and the ADPribose moiety. NAD+-consuming enzymes transfer ADPribose and/or ADPribose polymers, form signaling compounds from NAD+ and NADP, and reverse the acetyl modification of protein lysine residues. Each of these reactions consumes an NAD+ equivalent to a salvageable Nam product plus an ADPribosyl product.
 View larger version(19K) | Figure 1 NAD+ is both a coenzyme for hydride transfer enzymes and a substrate of NAD+-consuming enzymes. NAD+ is interconverted between oxidized and reduced states by NAD+/NADH-requiring hydride transfer enzymes. Both NAD+ and NADH can be phosphorylated by NAD+/NADH kinases. NADP and NADPH are interconverted by NADP/NADPH-requiring hydride transfer enzymes. These reactions are performed without net coenzyme consumption. In contrast, NAD+ is consumed to nicotinamide (Nam) and multiple forms of ADPribose by at least three types of NAD+-consuming enzymes. cADPribose synthases cyclize the ADP-ribose moiety of NAD+ and form NaADP from NADP with production of Nam. These enzymes also hydrolyze cADPribose and NaADP. ADPribosyltransferases (ARTs) post-translationally modify proteins with an ADPribosyl group, and polyADPribosyltransferases (PARPs) form a polymer of ADPribose from multiple NAD+ molecules. Sirtuins use the ADPribose moiety of NAD+ to accept the acetyl modification of a protein lysine, forming deacetylated protein plus Nam and acetylated ADPribose. NAD+ breakdown necessitates Nam salvage. Nicotinamide (Nam), nicotinic acid (Na), nicotinamide riboside (NR), and tryptophan (Trp) are the dietary precursors of NAD+. |
 View larger version(20K) | Figure 2 Intracellular NAD+ metabolism in humans. Tryptophan (Trp), nicotinic acid (Na), nicotinamide (Nam), nicotinamide riboside (NR), and possibly nicotinic acid riboside (NaR) are utilized through distinct metabolic pathways to form NAD+. Tryptophan (Trp) is converted to NAD+ in the eight-step de novo pathway. First, Trp is converted to N-formylkynurenine by the gene products of either indoleamine dioxygenase (INDO) or tryptophan dioxygenase (TDO2). The product of the arylformamidase gene (AFMID) forms kynurenine (Kyn) from N-formylkynurenine. Kynurenine is used as a substrate of kynurenine monooxygenase (KMO) and forms 3-hydroxykynurenine (3-HK). Kynureninase (KYNU) then forms 3-hydroxyanthranilate (3-HAA), which is converted to 2-amino-3-carboxymuconate semialdehyde (not shown) by 3-hydroxyanthranilate dioxygenase (HAAO). The semialdehyde undergoes a spontaneous condensation and rearrangement to form quinolinate (Quin), which is converted to nicotinic acid mononucleotide (NaMN) by quinolinate phosphoribosyltransferase (QPRT). NaMN is then adenylylated by the products of the NMNAT1-3 genes to form nicotinic acid adenine dinucleotide (NaAD+), which is converted to NAD+ by glutamine-dependent NAD+ synthetase (NADSYN1). Nicotinic acid (Na) is utilized in the three-step Preiss-Handler pathway. Nicotinic acid phosphoribosyltransferase (NAPRT1) forms NaMN by addition of the 5-phosphoribose group from 5-phosphoribosyl-1-pyrophosphate to Na. In two steps shared with the de novo pathway, NaMN is then converted to NaAD+ and NAD+ via activity of NMNAT1-3 and NADSYN1. Nam is utilized via nicotinamide phosphoribosyltransferase (Nampt), encoded by the PBEF1 (NAMPT) gene. Nampt catalyzes the addition of a phosphoribose moiety onto Nam to form nicotinamide mononucleotide (NMN). NMN is subsequently converted to NAD+ by the products of NMNAT1-3. Nam is produced by NAD+-consuming enzymes. Nam can be converted to Na by bacterial nicotinamidase in the gut for subsequent Preiss-Handler salvage. Nicotinamide riboside (NR) is phosphorylated by the products of nicotinamide riboside kinase genes (NRK1 and NRK2) to form NMN, which is converted to NAD+ by NMNAT1-3. NR may also be utilized by the product of the NP gene, purine nucleoside phosphorylase, for subsequent Nam salvage. NaR is utilized in yeast via the Nrk and NP pathways. The enzymes of human NAD+ biosynthesis are summarized in Table 1. |
|

NAD+-consuming enzymatic activities are induced, in part, by stresses such as DNA damage and inflammation. Many of these stresses are accompanied by specifically induced biosynthetic pathways, which appear to function to maintain NAD+ homeostasis. The term NAD+ homeostasis should be used cautiously, however, because it is not clear that cells are always in NAD+ homeostasis. Mammalian NAD+ biosynthesis is not a closed, cell-autonomous system, and there appear to be situations in which cells actively increase and/or reduce the concentration of NAD+ and NAD+ metabolites to promote vital and/or regulatory functions, including cell death.
| RECOMMENDED DAILY REQUIREMENTS OF THE NAD+ PRECURSORS |
|---|
Despite the fact that the biosynthetic pathways are not the same, in the literature of human and animal nutrition, Nam and Na are collectively termed niacin and/or vitamin B3. To protect against pellagra that can develop with Trp deficiency, recommended daily allowances (RDAs) of niacin are 16 and 14 mg per day for adult men and women, respectively . Because all plant, animal, and fungal inputs in our diet contain cellular NAD+ and NAD+ metabolites, foods provide NAD+, NADH, NADP, and NADPH, which are considered nutritional “niacin equivalents,” in addition to Nam and Na. Whereas Nam and Na are the fully broken down NAD+ metabolites from animals and plants/fungi/bacteria respectively, NR and nicotinic acid riboside (NaR) can be considered partly broken down niacin equivalents. In the genetically tractable yeast system, all of the salvageable precursors (Na, Nam, NR, and NaR) support the growth of cells inactivated for de novo NAD+ synthesis.
A single yeast cell deficient in de novo synthesis or undergoing a biological process that requires more than the minimum vital concentration of NAD+ must convert an available vitamin precursor to NAD+ in a cell-autonomous fashion. In contrast, humans exhibit the complexity of systemic NAD+ metabolism in which particular cells may utilize an NAD+ precursor to produce an excess of NAD+ and export salvageable precursors to other cells. Accordingly, dietary Trp is also classified as a niacin equivalent. However, because of the protein and other biosynthetic uses of Trp, 60 mg of Trp is considered the equivalent of 1 mg of niacin. This physiological fact, that high levels of dietary Trp result in circulation and excretion of Na , has resulted in claims in textbooks and reviews that Na is derived from Trp. Whereas this is true in vertebrate organisms, there may not be a vertebrate intracellular pathway that is responsible. As illustrated in Figure 2, Trp can be converted to Nam but not Na in any vertebrate cell expressing a de novo pathway and an NAD+-consuming enzyme such as poly(ADPribose)polymerase (PARP) or a sirtuin. Nam can then be converted in the intestinal lumen by bacterial nicotinamidase to Na. The most reasonable pathway by which a single vertebrate cell might convert Trp to Na was depicted by Magni and coworkers. Magni's scheme of human NAD+ metabolism depicts the activity of Na phosphoribosyltransferase (NAPRT1 gene product) and several other enzymes as bidirectional. Indeed, if there is sufficient pyrophosphate and NaMN in cells, then Naprt1 could catabolize nicotinic acid mononucleotide (NaMN) to Na, thereby creating a cell-autonomous route from Trp to Na. This is an interesting possibility that has not been demonstrated in vivo.
NR is a newly discovered salvageable precursor of NAD+ that occurs in cow's milk. Studies in Saccharomyces cerevisiae have shown that, like Na and Nam, NR is an NAD+ precursor that contributes to maintaining intracellular NAD+ concentration and improves NAD+-dependent activities in the cell including Sir2-dependent gene silencing and longevity. NR can either be converted to NAD+ by the Nrk pathway , which is induced by axotomy in dorsal root ganglion (DRG) neurons, or by the action of nucleoside phosphorylase and nicotinamide salvage. It has also been shown that the same two pathways required for NR salvage in yeast cells can also be used for NaR salvage. In yeast cells, NR clearly qualifies as a vitamin by virtue of rescuing growth of strains deficient in de novo synthesis , improving Sir2 functions, and utilizing a dedicated transporter. Additionally, because cells deleted for the NR/NaR salvage enzymes have a significant deficiency in intracellular NAD+ when not supplemented with these compounds, it appears that NR and/or NaR are also normal metabolites.
Five lines of reasoning support designation of NR as an authentic NAD+ precursor vitamin in vertebrates. First, Haemophilus influenza, a flu-causing bacterium, which has no de novo pathway and cannot utilize Na or Nam, is strictly dependent on NR, NMN, or NAD+ for growth in the host bloodstream. Second, milk is a source of NR. Third, NR protects murine DRG neurons in an ex vivo axonopathy assay via transcriptional induction of the nicotinamide riboside kinase (NRK) 2 gene. Fourth, exogenously added NR and derivatives increase NAD+ accumulation in a dose-dependent fashion in human cell lines. Fifth, Candida glabrata, an opportunistic fungus that depends on NAD+ precursor vitamins for growth, utilizes NR during disseminated infection.
It should be realized that not every cell is capable of converting each NAD+ precursor to NAD+ at all times. Expression of the eight-step de novo pathway is required to utilize trp. Expression of the Nampt pathway is required to utilize Nam. Expression of either the Nrk pathway or nucleoside phosphorylase and the Nampt pathway is required to utilize NR. Finally, expression of the Preiss-Handler pathway is required to utilize Na. Because tissue and cell-type specific enzyme expression differences exist, the precursors are differentially utilized in the gut, brain, blood, and organs. Understanding the unique aspects of metabolism of each precursor is necessary to define the mechanisms underlying the physiological effects and side effects of each.
| NAD+ FUNCTIONS AND THE REQUIREMENT FOR NAM SALVAGE |
|---|
NAD+ is classically known as a coenzyme for hydride-transfer enzymes. As a coenzyme, NAD+ is essential to a variety of diverse biological processes including energy production and synthesis of fatty acids, cholesterol, and steroids. NAD+ participates in oxidation-reduction (redox) reactions as hydride donor (NADH and NADPH) and acceptor (NAD+ and NADP). NAD+ most commonly functions in energy-producing catabolic reactions, such as the degradation of carbohydrates, fats, proteins, and alcohol, whereas NADP functions in anabolic reactions, such as the synthesis of cellular macromolecules including fatty acids and cholesterol. As depicted in Figure 1, coenzymatic activities of NAD+ and its reduced and phosphorylated derivatives interconvert but do not alter total cellular levels of NAD+.
In recent years, multiple enzyme-mediated, nonredox roles for NAD+ have been discovered. NAD+-consuming enzymes break down NAD+ to Nam and an ADPribosyl product. These enzymes fall into three classes. The first class consists of ADPribose transferases (ARTs) and PARPs, which transfer and/or polymerize NAD+-derived ADPribose, frequently as a post-translational modification. PARP activity, which is upregulated by DNA strand breaks, may be the major source of intracellular NAD+ consumption. The second class of NAD+-consuming enzymes consists of cADPribose synthases, which are membrane-bound ecto-enzymes also known as CD38 and CD157, that produce and hydrolyze the Ca2+-mobilizing second messenger cADPribose from NAD+. Additionally, CD38 catalyzes a base exchange between NADP and Na to form nicotinic acid adenine dinucleotide phosphate (NaADP), which is also a hydrolytic substrate. The products of these reactions have distinct and important roles in Ca2+ mobilization and signaling. The third class of NAD+-consuming enzymes consists of sirtuins, named for their sequence similarity to the yeast Sir2 gene-silencing protein. Sirtuins exhibit protein lysine deacetylase and, occasionally, ADPribose transferase activities. Sirtuin deacetylation reactions proceed by binding an acetyl-modified lysine on a target protein and NAD+ in distinct pockets. Deacetylation of the modified lysine side chain is coupled to the cleavage of the glycosidic bond in NAD+ such that the products are the deacetylated lysine, acetylated ADPribose, and Nam. Sirtuin-dependent deacetylation of histones and other proteins results in reprogrammed gene expression, mitochondrial synthesis and function, cell survival, and longevity. Sirtuins have been recently reviewed as master switches of metabolism.
Nam regulates the activity of NAD+-consuming enzymes both by direct enzyme inhibition and by its role as an NAD+ precursor. Nam inhibits NAD+-consuming enzymes by binding a conserved pocket that participates in NAD+ binding and catalysis. Recycling of Nam back to NAD+ raises NAD+ levels, increasing substrate availability and relieving Nam inhibition. In systems such as yeast in which NAD+ concentration has been determined carefully, basal intracellular NAD+ concentration is approximately 0.8 mM and can be elevated with vitamin precursors by 1 mM or more. At first glance, it would seem that 0.8 mM NAD+ should be saturating for virtually all sirtuins, which exhibit Km values for NAD+ between 5 μM and 500 μM. However, the first 0.8–1 mM of intracellular NAD+ concentration may largely be bound by redox enzymes such that salvage-derived synthesis of NAD+ may generate the majority of free NAD+ to drive sirtuin functions.
The first step in Nam salvage is catalyzed by Nam phosphoribosyltransferase (Nampt), which is encoded by the PBEF1 gene. This polypeptide, first identified as an extracellular protein termed pre B-cell colony enhancing factor (PBEF), was characterized as cytokine that enhances the maturation of B-cell precursors in the presence of IL-7 and stem cell factor. The same polypeptide was also termed Visfatin, due to reported insulin mimetic functions and regulation of systemic metabolism. However, the intracellular form of the molecule was shown to be induced in activated lymphocytes and function simply as Nampt.
Because of the disparate functions ascribed to the same polypeptide, it had been assumed that intracellular Nampt functions as an NAD+ biosynthetic enzyme, whereas extracellular Nampt has cytokine and insulin-mimetic roles. However, it has now been reported that both intra- and extracellular Nampt exhibit robust phosphoribosyltransferase activities and that inhibiting NAD+ biosynthesis through the Nampt pathway, either genetically or pharmacologically, causes impaired glucose tolerance and reduced insulin secretion in mice, a defect that can be corrected by administering nicotinamide mononucleotide (NMN). Moreover, murine plasma contains high concentrations of Nampt and NMN, which are reduced in nampt heterozygous animals. These new findings strongly indicate that the primary role for extracellular PBEF/Visfatin/Nampt is to catalyze NMN production from Nam, and that NMN has an important role in maintaining β-cell function. In a recent review, we speculated that a partially extracellular NAD+ cycle might consist of a Nampt step, followed by extracellular dephosphorylation of NMN to NR, intracellular transport of NR, and conversion of NR to NAD+.
| NAD+ PRECURSORS IN FOODS |
|---|
Consistent with Goldberger's studies, niacin is abundant in meat, eggs, fish, dairy, some vegetables, and whole wheat. Notably, corn contains abundant Na and Nam, largely present in bound forms that are not bioavailable. Treatment with alkali is used to increase bioavailability, a practice that protected native and South American populations from deficiency. Untreated corn is considered “pellagragenic,” causing increased sensitivity to low dietary niacin concentrations in animal studies. Milk, now known to be a natural source of NR, was shown to counteract the growth defect seen in corn-fed animals. In meats, Na and Nam are scarce and NAD+ and NADP are the abundant sources of niacin. Nam is produced by mucosal enzymes that cleave NAD+ , and Na is produced from Nam by deamination by bacterial nicotinamidase in the gut. Both Na and Nam are absorbed from the alimentary canal and enter the bloodstream for distribution to tissues. Studies indicate that Nam is the dominant absorbed form of niacin when the dietary sources are NAD+ and NADP. However, it has also been reported that NAD+ is digested by pyrophosphatases to NMN and hydrolyzed to NR, which was found in the lumen of the upper small intestine. We surmise that NR is incorporated into the cellular NAD+ pool via the action of Nrk pathway or via Nam salvage after conversion to Nam by phosphorolysis.
| SYSTEMS ANALYSIS OF NAD+ BIOSYNTHESIS |
|---|
Qprt, Nampt, Naprt1, and Nrk1,2 are the committed enzymes in the synthesis of NAD+ from Trp, Nam, Na, and NR. As such, by examining the expression of each enzyme and by following the metabolic fates of dietary inputs, one can describe tissue-specific pathways of NAD+ biosynthesis.
Animals on diets containing sufficient amounts of both Trp and niacin have a measurable concentration of each in liver. Nam and Na, however, are thought to supply only a fraction of the NAD+ produced in the liver, with much niacin circulating to other tissues. Trp is thought to be the principal NAD+ precursor utilized in liver. In addition to producing quinolinate for entry into NAD+ biosynthetic pathways, Trp is incorporated into protein, utilized to generate energy through total oxidation, and utilized to form kynurenic acid. Inducible enzymes of Trp utilization regulate the flux of Trp through different pathways depending on diet and cellular metabolic state. Under conditions of low Trp consumption, circulating levels of Trp decrease and enzymes that direct Trp to non-NAD+ biosynthetic routes are down-regulated, suggesting a shift of all possible Trp catabolism to NAD+ generation. Supplementing high amounts of Trp allows more flux to the oxidative branch and allows increased levels of Nam to be released into the vasculature. All Trp that reaches quinolinate in the liver is thought to be converted to NAD+ via subsequent enzyme reactions of Qprt, Nmnat1-3, and Nadsyn1, which are highly expressed in liver.
In addition to liver, Qprt is expressed in human and rat brain and plays a critical role in protection against the neurotoxic effects of quinolinate. Quinolinate is a potent endogenous neurotoxin, and elevated levels in brain are associated with neurodegenerative disorders including epilepsy and Huntington's disease. The normal concentration of quinolinate in the brain was found to be in the low- to mid-nanomolar range, and Qprt activity increases in response to increased levels of quinolinate, suggesting a protective role. The highest levels of quinolinate are found in spleen, lymph nodes, thymus, and many specific immune cell types and are increased following stimulation by immune activators.
Activated lymphocytes induce expression of Nampt, which is also expressed in smooth muscle cells with loss of expression in senescence. In the mouse, Nampt has been shown to circulate and to be highly expressed intracellularly in brown adipose tissue, liver, and kidney, with fat as the source of extracellular Nampt. Human fat is also a source of circulating Nampt. From classical feeding studies, the testes were found to utilize Nam rather than Na or quinolinate, and blood and liver were also found to be major sites of Nam utilization. Nam crosses the blood-brain barrier and is converted to NAD+ in brain, though it is not known whether Nam is a precursor of NAD+ in neuronal or non-neuronal cells. Studies with DRG neurons suggest that Nampt is not neuronally expressed. Nam and NR are also taken up by intestinal epithelial cells and both are utilized by Nam salvage.
By global analysis of mRNAs, Naprt1 is expressed in most tissues of the adult mouse, including colon, heart, kidney, and liver, suggesting the presence and utilization of the substrate Na as an NAD+ precursor in these tissues. Classical feeding studies showed that exogenously added Na is a better NAD+ precursor than Nam in liver, intestine, and kidney. Similarly, rats fed Na showed elevated levels of NAD+ in the heart and kidney in addition to blood and liver, which are sites of Na and Nam utilization. Classical studies have been corroborated by a recent report that mouse Naprt1 is expressed in intestine, liver, kidney, and heart. In addition, human kidney cell lines are able to use Na to increase intracellular NAD+ concentration in a manner that depends on the NAPRT1 gene. Moreover, Naprt1 expression decreases vulnerability to oxidative damage from NAD+ depletion. The use of Na as an NAD+ precursor in normal and stress conditions implicates the presence of Na as a normal cellular metabolite in humans. As discussed earlier, we consider the bacterial flora of the intestinal lumen to be the first major site in a vertebrate for production of Na, though bacterial and fungal degradation of cellular NAD+ in food and direct Na supplementation will also produce supplies of Na in the alimentary canal for distribution to tissues through the vasculature. True intracellular production of Na in a vertebrate cell would require high levels of Trp and/or NAD+ and substantial reverse flux through what are usually considered anabolic pathways.
The use of NR as a precursor in mammalian cell types was first demonstrated in DRG neurons, which induce the NRK2 transcript when damaged by axotomy. The ubiquitous expression of Nrk1 in mammalian tissues suggests utilization of NR and/or NaR in a diverse array of cell types. However, Nrk2 is present in heart, brain, and skeletal muscle, and is notably absent in kidney, liver, lung, pancreas, and placenta . The fact that DRG neurons cannot be protected from damage-induced neuropathy by Na or Nam without concurrent gene expression of Na or Nam salvage genes suggests that NR is a uniquely useful precursor to the nervous system when de novo synthesis of NAD+ from Trp is not sufficient.
Available data summarizing the system-wide use of NAD+ precursors are summarized in Figure 3. The data are more static than one would like, such that it will be important to determine how gene expression and precursor utilization changes as a function of nutrition, age, stress, and disease state. It is particularly striking that two enzymes, namely Nampt and Nrk2, were first identified as highly regulated proteins involved in immune cell and muscle cell development. Thus, developmental regulation of NAD+ synthesis and utilization remains on the forefront of NAD+ biology.
 View larger version(56K) | Figure 3 Tissue specificity of nicotinamide adenine dinucleotide (NAD+) metabolism. Tissue distributions for key pathway enzymes Qprt, Nampt, Naprt1, and Nrk1/2 are represented by pathway identifiers De novo, Nam salvage, Preiss-Handler, and Nrk pathway, respectively. Food is broken down in the alimentary canal to NAD(H), NADP(H), Nam, Na, NR, and Trp. Salvageable precursors Nam, Na, and NR are absorbed by intestinal epithelia and transported to the bloodstream. Mucosal enzymes break NAD(H) and NADP(H) into Nam and NR, and bacterial nicotinamidase in the intestinal lumen produces Na from Nam. The intestine conducts Preiss-Handler and Nam salvage. Circulating precursors are taken up by various tissues. The de novo pathway is active in liver, neuronal, and immune cells. Intracellular Nam salvage is active in adipose tissue, liver and kidney, and immune cells in addition to intestine. Extracellular Nampt is produced by adipocytes and circulates in plasma. Naprt1 is most highly expressed in liver, kidney, and heart in addition to the intestine. The Nrk pathway is expressed in neurons and in cardiac and skeletal muscle. |
| ALTERATION OF NAD+ METABOLISM BY CALORIC RESTRICTION |
|---|
Caloric restriction (CR) is the most effective intervention to extend the lifespan of multiple model organisms including mammals. CR is defined as a 20% reduction versus ad libitum feeding without compromising adequate nutrition. Although the mechanisms of CR remain elusive, it is thought that CR modulates fat and carbohydrate metabolism, attenuates oxidative damage, and activates a stress-induced hormetic response that mediates improved vitality and disease resistance. Among these three major mechanisms, modulation of fat and carbohydrate utilization is the most direct response to reduced dietary inputs, and hormesis is potentially the mechanism most influenced by the “signaling” aspect of CR.
The hormetic theory is supported by experiments in which model organisms exhibit extended lifespan when placed in a variety of sublethal stress conditions including high temperature, high salt, or osmotic stress. In yeast, such conditions increase expression of nicotinamidase, thereby altering NAD+ metabolism in a manner that favors Sir2 activity. The principal mechanism by which Sir2 extends lifespan in a wild-type yeast cell is repression of formation of aging-associated extrachromosomal ribosomal DNA circles. Though this mechanism is unique to yeast, there are substantial data showing that sirtuins are conserved from fungi to metazoans to mediate some of the beneficial effects of CR. Sir2 is not the only mediator of CR-induced lifespan extension in yeast, nor are sirtuins necessarily the only targets of nicotinamide inhibition. Nonetheless, there is excellent evidence that NAD+ metabolism is altered in vertebrate systems by CR and that increased activity of sirtuins may mediate beneficial brain and liver physiology under CR conditions.
In CR-treated mice, brain NAD+ levels are increased and Nam levels are decreased, and these changes accompany neuronal Sirt1 activation, which reduces Alzheimer's neuropathology. In fasted mice, NAD+ levels are increased in liver, which is accompanied by Sirt1 activation, PGC1α deacetylation, and increased mitochondrial biogenesis. The mechanisms by which lower food inputs increase NAD+ levels in brain and liver are completely unknown. Two potential mechanisms that may account for this phenomenon are systemic mobilization of NAD+ precursors to the brain and liver and reduced NAD+ breakdown. Among the potential precursors that could mediate this phenomenon, Na and Trp seem unlikely because one would expect that increased food consumption would be required to increase their availability. Analysis of CR-induced systemic metabolites should permit the detection of either Nam or NR as candidate mediators of increased brain and liver NAD+ levels. Reduced NAD+ breakdown is another mechanism by which CR might increase NAD+ levels in particular tissues. This could occur if an NAD+-consuming activity such as PARP is negatively regulated by CR. The pathways that produce NR or NaR at the expense of NAD+ are not known. These, too, might be negatively regulated by CR in order to elevate brain and liver NAD+.
| LESSONS FROM THE wlds MOUSE |
|---|
Wallerian degeneration refers to the ordered process of axonal degeneration. Wallerian degeneration occurs when an axon is severed from the cell body, and proceeds via characteristic fragmentation of cellular components initiated by a factor or factors intrinsic to the neuron. The distal part of a severed or damaged axon usually undergoes Wallerian degeneration within 24–48 hours of injury. This type of axonopathy is thought to be a critical, early event in neurodegenerative conditions including multiple sclerosis, Alzheimer's, and Parkinson's diseases and in polyneuropathies associated with diabetes and acute chemotherapy use. Remarkably, a mouse mutant, termed wlds, with delayed Wallerian degeneration has been identified in the C57BL/6 background. Axons from the wlds mouse survive several weeks after transection.
The dominant neuroprotective gene in the wlds mouse is an in-frame fusion of the N-terminal 70 amino acids of a ubiquitin assembly factor (Ube4b/Uf2a) with the entire coding sequence of Nmnat1. Transgenic mice expressing the Ube4b/Nmnat fusion gene showed that the nuclear protein protected from axonopathy in a manner that depended on the level of protein expression, indicating the activity of a nuclear-derived factor. Although Nmnat in flies can protect against degeneration of optic neurons in an active site-independent manner, the protective factor for DRG neurons appears simply to be NAD+. The evidence is as follows. Lentiviral expression of Nmnat1 protects DRG neurons from axonopathy in an active site-dependent manner. Overexpression of wlds or Nmnat1 prevents NAD+ and ATP decline in response to mechanical and chemical damage. Nam and Na also protect against axonopathy as long as Nampt or Naprt1 are concomitantly expressed in DRG neurons, whereas NR protects without engineered gene expression of a biosynthetic gene. Nrk2 mRNA levels following axonopathy are induced approximately 20-fold, indicating a preferential use of NR as a precursor in maintaining intracellular NAD+ levels in DRG neurons. Whether sufficient oral NR supplementation might protect against diabetic or chemotherapy-induced neuropathy or protect against age-associated neurodegenerative conditions remains to be determined.
| NICOTINIC ACID MECHANISMS OF ACTION IN DYSLIPIDEMIA |
|---|
Na has been used to treat dyslipidemias in humans since the 1950s. Gram dosages reduce triglycerides and low-density lipoprotein (LDL, i.e., “bad”) cholesterol and raise high-density lipoprotein (HDL, i.e., “good”) cholesterol levels. As a monotherapy, Na is one of the most effective means to improve cardiovascular risk factors and, in combination with statins and bile acid treatments, can enhance therapeutic effects. In addition to lowering circulating cholesterol levels, Na prevents establishment of lipid deposits and the progression of atherosclerosis in a cholesterol-fed rabbit model. However, high-dose Na utilization produces a painful flushing response that limits use.
The mechanism of action of Na in treatment of dyslipidemias is not clear. Na is an agonist of the G-protein-coupled receptor Gpr109A (PUMAG in mice). However, it is not clear that activation of this receptor, which is not expressed in the liver, can account for the clinical efficacy of Na. Activation of Gpr109A in adipocytes inhibits the liberation of free fatty acids from stored triglycerides. However, activation of GPR109A in epidermal Langerhans cells is directly responsible for flushing. The lack of expression of Gpr109A in the liver and the finding that Gpr109A mediates flushing cast serious doubt on the receptor model of Na function in dyslipidemia.
We have hypothesized that the beneficial effects of high-dose Na derive simply from NAD+ biosynthesis. The fact that Nam is not beneficial in promoting reverse cholesterol transport can be explained in two ways. First, Na is a better NAD+ precursor than Nam in liver. Second, if the requirement for elevated NAD+ biosynthesis for improved reverse cholesterol transport depends on sirtuin function, one would expect Nam to be inhibitory.
Sirt1 has been identified as a positive regulator of liver X receptor (LXR), which in turn is a regulator of cholesterol and lipid homeostasis. Sirt1 deacetylates LXR at conserved lysine residues, resulting in LXR activation. sirt1−/− animals show reduced expression of LXR target genes, SREBP1, and ABCA1, in macrophages and in liver, which play important roles in HDL biogenesis. These data would appear to make the simple model of Na as an NAD+ precursor highly reasonable.
O artigo completo está nesse link: http://arjournals.annualreviews.org/doi/full/10.1146/annurev.nutr.28.061807.155443?
Por: Pollana Roberta Alves Campos
Assinar:
Comentários (Atom)
